Administración
Nacional de Medicamentos, Alimentos y Tecnología Médica
ESPECIALIDADES
MEDICINALES
Disposición
705/2005
Requisitos para
la inscripción de vacunas. Presentación de la documentación técnica.
Establecimientos elaboradores. Información Preclínica-Clínica. Formularios.
Bs. As., 7/2/2005
VISTO el
Expediente Nº 1-0047-1110-3469-04-4 del Registro de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, y
CONSIDERANDO:
Que las
actividades de elaboración, producción, fraccionamiento, importación,
exportación y depósito en jurisdicción nacional o con destino al comercio
interprovincial de medicamentos se encuentran regidas por la Ley 16.463 y los Decretos 9763/94, 150/92 y sus modificatorios Nº 1890/92 y 177/93 y normas
complementarias.
Que en el marco
descripto las especialidades medicinales requieren para su inclusión en el
Registro de Especialidades Medicinales de un proceso de evaluación de la
documentación que dé cuenta de la seguridad y eficacia del producto de que se
trate.
Que las vacunas
que previenen enfermedades pueden tener una complejidad y características
propias para cada tipo de producto, y son de aplicación en población sana.
Que la Organización Mundial de la Salud (OMS) define a las vacunas preventivas como las
preparaciones que contienen sustancias antigénicas capaces de inducir en el
hombre una inmunidad activa y específica contra un agente infeccioso, sus
toxinas o antígenos elaborados por él y ha formulado recomendaciones a todas
las agencias regulatorias con el fin de garantizar la utilización de vacunas de
calidad ("Reglamentación de Vacunas: desarrollo en los organismos actuales
de reglamentación farmacéutica" (WHO/V&B/99.10).
Que en particular,
respecto a esta Administración Nacional, los expertos de la OMS y de la Oficina Sanitaria Panamericana, luego del proceso de evaluación llevado a cabo en
nuestro país en marzo de 2000 y en agosto de 2003, efectuaron diversas
recomendaciones de las que surge la necesidad de optimizar los requerimientos
para el registro de las vacunas en la Argentina.
Que en este
contexto resulta conveniente especificar los requisitos científicos y técnicos
requeridos para acreditar en forma fehaciente la calidad, eficacia y seguridad
de las vacunas.
Que a esos efectos
se han tomado como referencia los requisitos mínimos recomendados por la Oficina Sanitaria Panamericana para el registro de vacunas en la región de Las Américas.
Que desde el punto
de vista operativo es conveniente adoptar la modalidad de gestión descripta de la Disposición ANMAT 5755/97, a cuyos fines se ha elaborado un formulario específico para la
tramitación de las solicitudes de inscripción de vacunas.
Que el Instituto
Nacional de Medicamentos, la Dirección de Evaluación de Medicamentos y la Dirección de Asuntos Jurídicos han tomado la intervención de su competencia.
Que se actúa en
virtud a las facultades conferidas por el Decreto Nº 1490/92 y el Decreto Nº
197/02.
Por ello,
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1º — La presente disposición se aplicará a las
tramitaciones de solicitudes de inscripción de vacunas, entendiéndose por tales
a las preparaciones que contienen sustancias antigénicas capaces de inducir en
el hombre una inmunidad activa y específica contra un agente infeccioso, sus
toxinas o los antígenos elaborados por éste.
Art. 2º — Apruébanse los requisitos y la documentación para la
tramitación de las solicitudes de inscripción de vacunas, los que como Anexo I
y Anexo II (Formulario 1.2.VAC) forman parte integrante de la presente
disposición.
Art. 3º — Esta Administración Nacional se expedirá respecto a
los trámites de inscripción de vacunas dentro de un plazo de CIENTO VEINTE
(120) DIAS HABILES ADMINISTRATIVOS.
Art. 4º — Establécese que las solicitudes de inscripción de
vacunas devengarán un arancel de CUATRO MIL PESOS ($ 4000).
Art. 5º — La presente disposición entrará en vigencia el día
siguiente al de su publicación en el Boletín Oficial.
Art. 6º — Regístrese; comuníquese a quienes corresponda.
Notifíquese a CAEME; CILFA, COOPERALA. Publíquese, dése a la Dirección Nacional del Registro Oficial. Cumplido, archívese PERMANENTE. — Manuel R. Limeres
ANEXO I
REQUISITOS PARA LA INSCRIPCION DE VACUNAS.
DOCUMENTACION
TECNICA
1. INFORMACION
SOBRE EL PRODUCTO
El fabricante debe
presentar información suficiente que demuestre la inocuidad, calidad y eficacia
del producto. La documentación presentada debe incluir los siguientes
elementos:
1. 1. - Proceso de
Producción:
Se deberá
describir el proceso de fabricación de la vacuna desde la materia prima de
origen biológico (banco de células, cepas semilla, etc.). El proceso de
producción tanto de los antígenos que componen la vacuna como del producto
final, debe ajustarse a las normativas establecidas por los Comités de Expertos
en Productos Biológicos de la Organización Mundial de la Salud, recogidas en la Serie de Informes Técnicos y, en los casos que corresponda, a las normas de
otras autoridades y organismos reguladores tales como Food and Drug
Administration (FDA/USA), Conferencia Internacional de Armonización (ICH) y
Agencia Europea de Evaluación de Medicamentos (EMEA).
Además, se deberá
precisar el nombre del laboratorio productor de cada antígeno y del producto
final indicando la responsabilidad del mismo. En el caso de ser laboratorios
diferentes, se incluirá la información relacionada a las medidas tomadas para
el traslado del material de un sitio al otro.
Durante la
producción se deberá cumplir con las Buenas Prácticas de Producción de
Productos Farmacéuticos establecidas por la ANMAT complementadas por las Buenas Prácticas de Fabricación de Productos Biológicos recomendadas por la OMS (SIT 822 y/o sus actualizaciones).
Se deberá incluir
la siguiente información:
- Fórmula Maestra
para cada tamaño de lote
- Descripción
detallada de los materiales de partida tales como cepas, línea(s) celular(es),
huevos embrionados, animales, microorganismos, etc. incluyendo sus
especificaciones de calidad, las técnicas de análisis utilizadas para su
control y detección de agentes adventicios. En el caso de vacunas obtenidas por
la tecnología del ADN recombinante, se aportará la información correspondiente
a la célula huésped transformada y los sistemas de expresión.
- Se deberá
remitir la información y diagrama de flujo de producción de cada uno de los
antígenos que componen la vacuna y del producto final, incluyendo el tamaño del
lote en cada una de las etapas.
- En los casos en
que corresponda deberá incluirse información sobre los pasos de fermentación,
cosecha, inactivación, conjugación y purificación incluyendo los criterios de
aceptación y rechazo.
- Identificación
de los pasos críticos de la fabricación.
- Criterios de
aceptación o rechazo de los reprocesos para cada etapa.
- Controles en
proceso: especificaciones de calidad que incluyan la caracterización y pureza
del producto obtenido en cada etapa y las técnicas analíticas utilizadas.
- Descripción de
los procedimientos de llenado y cierre para el producto final.
- Procedimientos
de acondicionamiento.
- Descripción de
los procedimientos de cadena de frío utilizados.
1.2. - Control de
calidad
- Control de
calidad de Materias Primas: especificaciones de calidad de todos los materiales
y solventes utilizados para la producción de la vacuna.
- Control de
Calidad de productos intermediarios: procedimientos analíticos con sus
correspondientes validaciones.
- Control del
producto final: procedimientos analíticos para el control de calidad del
producto final con sus correspondientes validaciones.
- Materiales de
referencia: describir las características de los materiales de referencia
utilizados en los controles de calidad en proceso y a nivel del producto final,
incluyendo cuando proceda, los estudios comparativos con el material de
referencia definido por la Autoridad regulatoria.
1.3. - Estudios de
estabilidad:
Se adjuntará el
informe de los estudios de estabilidad de graneles y producto final, de por lo
menos tres lotes, procedentes de materiales de partida diferentes, de acuerdo
con las condiciones de almacenamiento y envase propuesto, especificando los
métodos analíticos empleados. Estos estudios deberán avalar un período de
validez corno mínimo de 12 meses y las condiciones de almacenamiento propuestas
para la materia prima activa, los productos intermedios que requieran
almacenamiento y el producto final. Los informes deben contener el nombre de la
vacuna, número de lotes estudiados, forma farmacéutica, potencia por dosis o
unidad, condiciones de almacenamiento del estudio, características y tipo de
envase primario utilizado, métodos de análisis utilizados, especificaciones y
resultados.
Los informes deben
incluir los datos relacionados a la identificación de los lotes estudiados, la
fecha de fabricación de cada uno, la fecha en que se realizó cada análisis y
sus resultados. El informe final debe presentar las conclusiones del
solicitante en cuanto al período de validez del producto en las condiciones de
almacenamiento y envase propuesto.
Cuando se trate de
productos liofilizados se deberá presentar los informes correspondientes a los
estudios de estabilidad del reconstituido y, para envases multidosis, estudios
donde se evalúe la esterilidad del producto en condiciones de uso.
1.4. -
Consistencia de la producción
Debe evaluarse la
consistencia de producción tanto para los antígenos como para el producto final
a través de la presentación de los protocolos analíticos de tres a cinco lotes
consecutivos, utilizando distintos materiales de partida y un tamaño de lote
que se corresponda con los de la rutina de producción.
1.5. -
Validaciones
Debe incluirse el
Plan de Validación para los procesos, reprocesos y métodos analíticos no
codificados en Farmacopea. Durante la verificación de Buenas Prácticas de
Fabricación y Control de Calidad del primer lote de fabricación de la vacuna se
deberá presentar la documentación completa de Validación.
1.6. -
Certificados analíticos
Debe incluirse un
modelo del certificado analítico para los principios activos, sustancias
auxiliares, productos en proceso cuando corresponda, producto terminado y
materiales de referencia. Todos los certificados del producto deben incluir
nombre de la sustancia o producto, etapa de elaboración, identificación de
lote, fecha y lugar de fabricación, tamaño del lote, especificaciones de
calidad (físicos, químicos, biológicos y microbiológicos) con sus límites y
resultados, vencimiento cuando corresponda, fecha y lugar de realización del
ensayo, nombre y firma del analista y supervisión y conclusión final.
1.7. -
Procedimientos operativos estándar
Debe presentarse
el listado de procedimientos operativos estándar necesarios para los procesos
de producción y de control.
1.8. - Rótulos y
prospectos
Deben incluirse en
la presentación los proyectos de rótulos, etiquetas y prospectos que serán
incluidos en el envase final.
2. INFORMACION
SOBRE EL ESTABLECIMIENTO ELABORADOR
El fabricante debe
proporcionar información suficiente para demostrar que aplica los principios de
Buenas Prácticas de Fabricación, inclusive la existencia de un sistema adecuado
de calidad. Pueden utilizarse planos, diagramas, gráficos de los procedimientos
ordinarios de operación y textos en que se transmita la información necesaria
en relación con los siguientes aspectos:
• Personal:
Organización y jerarquía, planes de capacitación y sistema de mantenimiento de
archivos.
• Localización y
construcción de el/los edificio/s utilizado/s para fabricación y control.
• Circulación de
personal, materias primas, producto semielaborado y terminado en las
instalaciones.
• Instalaciones
para animales.
• Sistemas de
agua, aire y vapor y suministro de energía eléctrica.
• Sistemas de
drenaje y evacuación de efluentes.
• Procedimientos y
programas de limpieza y medidas de control.
• Procedimientos
de garantía y control de calidad.
• Procedimientos
de validación.
• Sistemas de
documentación y de mantenimiento de registros.
• Procedimientos
de retiro de productos del mercado y posterior recuperación.
3. - INFORMACION
PRECLINICA - CLINICA
A los fines de la
organización y requerimientos de la información preclínica y clínica se ha
considerado, que todas las vacunas que son sometidas a registro constituyén
para la autoridad Sanitaria una nueva vacuna de la cual se debe disponer de los
datos apropiados de seguridad y eficacia. En función del conocimiento previo de
las vacunas se establecen diferencias de requerimientos entre vacunas noveles o
innovativas de las vacunas tradicionales según definición en el glosario que
forma parte de la presente.
Los criterios
conceptuales de los estudios Preclínicos y Clínicos son los lineamientos de los
documentos aprobado por el Comité de Expertos de Sustancias Biológicas la OMS: Guía para evaluación no clínica de vacunas 2003 y Evaluación Clínica de Vacunas desde la
perspectiva regulatoria 2001.
Se deberá incluir
la siguiente información:
- Exposición
Sumaria: resumen de la justificación técnica, científica y sanitaria por la
cual el producto constituye una vacuna segura y eficaz y la justificación de la
inclusión de cada uno de los ingredientes activos, aditivos y excipientes en la
fórmula.
- Informe del
Experto: informe elaborado por uno o más expertos en el que describan los
aspectos químico-farmacéuticos, preclínicos y clínicos del producto.
- Los fundamentos
que justifiquen: Condiciones de administración: indicaciones, posología, vías
de administración y modo de uso que se incluirán en los prospectos.
- Las
informaciones de seguridad que justifiquen Restricciones de uso: advertencias,
precauciones, contraindicaciones, reacciones adversas e interacciones que se
incluirán en los prospectos.
3.1 Información
preclínica.
3.1.a. Vacunas
noveles
Para el caso de
vacunas noveles deberá presentarse estudios preclínicos que incluyan la
información correspondiente a criterios de selección del ensayo y parámetros de
calidad, justificación de la selección de la dosis y parámetros de evaluación,
descripción de los materiales y métodos, resultados, análisis estadístico,
conclusiones y bibliografía. Cada uno de estos estudios debe estar precedido
por un resumen de las investigaciones realizadas con un análisis de los
resultados más relevantes, acompañado de una tabla representativa. Esta tabla
debe contener información sobre: tipo de ensayo, duración, especie, vía de
administración, rangos de dosis y resultados más significativos. Se presentarán
resultados de los estudios realizados sobre las especies relevantes para el
hombre. Se determinará la aparición de anticuerpos y se presentará el análisis
de su relevancia para el ensayo en cuestión y para el hombre.
Los diseños
experimentales deben ajustarse a las normativas aceptadas o dictadas por el
Comité de Expertos de la Organización Mundial de la Salud, ICH, EMEA, y FDA y cumplir con Buenas Prácticas de Laboratorio (BPL).
Con excepción del
hidróxido, fosfato de aluminio y aquéllos ya en uso autorizados por la Autoridad Sanitaria, los nuevos adyuvantes deberán presentar la evaluación preclínica,
incluidos los efectos sobre el sistema inmunológico. Lo mismo se indica para el
caso de nuevos estabilizantes, conservadores o nuevos aditivos en general que
no hubieran sido utilizados para las vías de administración ya conocidas.
Los estudios sobre
efectos farmacodinámicos deben enfocar principalmente dos aspectos: las
características de la respuesta inmune y la capacidad de protección frente a
los organismos patógenos. Debe presentarse los datos sobre curva dosis-efecto y
ensayo de potencia en los casos que corresponda. Se presentarán estudios
comparativos con productos de referencia conocidos siempre que sea posible y se
presentarán además, cuando corresponda, la comparación con los materiales de
referencia apropiados. En los estudios de respuesta inmunogénica de las vacunas
se presentarán datos sobre la cinética de aparición de los anticuerpos, media
geométrica, inmunidad mediada por células, duración de la respuesta inmune y
esquema de vacunación que justifiquen los esquemas propuestos para el humano.
En el caso de vacunas ADN, se presentará los resultados de esta evaluación en
relación a la generación de antígeno.
Se incluirá
documentación acerca de ensayos de provocación que demuestren la protección
inducida por la vacuna. Si la vacuna está destinada a la protección del recién
nacido se presentarán ensayos realizados en animales gestantes, que evalúen la
respuesta inmunológica del feto y el nivel de protección frente al estudio de
provocación.
Para el caso de
vacunas noveles se presentarán resultados de estudios in vivo e in vitro que
evalúen las acciones secundarias del producto sobre los órganos y sistemas del
organismo, especialmente sistema cardiovascular y respiratorio.
Los estudios
farmacocinéticos deberán tenerse en cuenta en el caso de nuevas formulaciones
y/ o vías de administración alternativas (oral o intranasal).
Los estudios de
toxicidad deberán incluir:
• Toxicidad por
administración única: se presentarán como mínimo estudios realizados en una
especie de mamíferos (con identificación de origen conocido) con una dosis que
aporte un adecuado margen de seguridad. Entre las vías de administración se
incluirá la propuesta para utilizar en humanos. La duración del período de
observación deberá considerar la vida media y las características del producto.
• Toxicidad por
administración reiterada: Son requeridos para las vacunas que requieren
múltiples dosis en el humano. Deberán evaluarse reacciones de
hipersensibilidad, inmunotoxicología y cuando sea necesario considerar la
posibilidad de reactividad cruzada con tejidos humanos normales (por ejemplo:
vacunas de ADN).
En el caso de las
vacunas de ADN se presentarán resultados de la investigación de manifestaciones
de autoinmunidad, inmunosupresión, tolerancia y presencia de anticuerpos
(anti-ADN, anti-lgG y otros). Este estudio puede ser parte de los ensayos de
Inmunogenicidad o de los estudios farmacológicos secundarios.
En el caso de
vacunas que se pretendan administrar a embarazadas, se presentará los
resultados de la evaluación de la seguridad del feto frente a la administración
de la vacuna y la respuesta inmunológica producida.
Para vacunas que
pueden ser utilizadas en mujeres en edad reproductiva el riesgo de toxicidad
embriofetal y perinatal debe ser razonablemente excluido mediante documentación
clínica y/o epidemiológica con datos sobre exposición al agente patógeno o
vacunas relacionadas. En otros casos, debe considerarse la disponibilidad de
modelos animales apropiados. Estos estudios también serán requeridos en el caso
de nuevos adyuvantes, nuevos agentes conservadores, estabilizadores u otros
aditivos.
Los estudios de
potencial mutagénico serán requeridos para vacunas de ADN. También serán
requeridos en el caso de nuevos agentes conservadores, estabilizadores u otros
aditivos.
Los ensayos de
oncogenicidad in Vivo e in Vitro serán necesarios en los casos de vacunas de
ADN, cuando exista integración al ADN del huésped o extensa homología con el
genoma humano, amplia distribución tisular o cuando el vector comprenda
secuencias con potencial oncogénico o se pretende aplicar en forma reiterada en
el tratamiento de enfermedades que no implican grave riesgo para la vida.
También serán requeridos en el caso de nuevos agentes conservadores,
estabilizadores u otros aditivos.
Deberá evaluarse
la tolerancia local de la formulación que se pretende emplear en el humano. Los
efectos locales potenciales pueden ser evaluados a partir de los ensayos de
toxicidad por administración única y reiterada.
3.1.b. Vacunas
convencionales:
Para el caso de
vacunas convencionales, deberá presentarse la información bibliográfica que
respalde la información farmacodinámica o de seguridad para la formulación
objeto de la solicitud de registro.
3.2 Información
Clínica
3.2.a. Vacunas
noveles:
Se requiere la
presentación de estudios clínicos de Fase I, II y III.
Los estudios
clínicos deberán estar dirigidos a la evaluación de la inmunogenicidad,
seguridad y eficacia de la vacuna en estudios Fase I, II y III. La información
deberá incluir la descripción detallada de los principales aspectos del
protocolo y los métodos analíticos utilizados, haciendo énfasis en:
– Diseño de la
investigación y cumplimiento de las Buenas Prácticas Clínicas
– Características
de la población estudiada
– Resultados
– Evaluación
estadística de los resultados.
– Presentación de
resultados clínicos y de laboratorio
– Discusión y
conclusiones
– Estudios
Farmacológicos Fase I y II que evalúen:
Farmacocinética:
Estudios que evalúen la farmacocinética en seres humanos en el caso de nuevas
vacunas conjugadas, nuevas vías de administración, vacunas de ADN,
estabilizadores, u otros aditivos.
Farmacodinamia:
Dirigidos a estudiar la actividad de la vacuna, incluyendo título, clase,
subclase y función de los anticuerpos específicos producidos, evaluación de la
lnmunogenicidad dosis- respuesta, tiempo de aparición y duración de adecuados
títulos de anticuerpos, inducción de la inmunidad celular, formación de
inmunocomplejos o cualquier interacción que pueda afectar el sistema
inmunológico. Estos estudios serán la base para la dosis recomendada en
relación con la cantidad de antígeno, número e intervalo de tiempo entre las
dosis de vacunación y posible necesidad de refuerzos.
En el caso de
nuevas vacunas combinadas se deberá evaluar la posible interferencia
inmunológica entre los antígenos, los efectos adversos en relación con la
administración individual de los antígenos y la posible interferencia con
vacunas cuya administración esté recomendada en el mismo período de tiempo en
una población en particular.
Los datos de
seguridad deberán estar relacionados con la administración de dosis simples y
repetidas, tanto en el ámbito local en el sitio de la administración como
reacciones sistémicas.
Estudios de
Eficacia (Fase II y III): La eficacia de la vacuna deberá evaluarse en estudios
randomizados controlados realizados en la población susceptible a la vacunación.
Es recomendable que se inicien con la evaluación pre-exposición o con
suficientes datos epidemiológicos de la enfermedad. En el caso de estudios no
controlados deberán justificar las razones de su desarrollo.
Para el caso de
nuevas vacunas que contengan antígenos para los cuales se encuentren
establecidos los niveles de anticuerpos protectores, los estudios de
inmunogenicidad se consideraran para establecer su eficacia.
Ei desarrollo de
otros tipos de estudios clínicos, como estudios prospectivos fase III, estudios
caso-control, estudios cohorte observación, serán evaluados según los
antecedentes del disponibles para el tipo de antígeno.
Los ensayos
clínicos de vacunas combinadas deben incluir la evaluación frente a los
antígenos administrados de manera individual y demostrar las ventajas
terapéuticas de la asociación, deberán asimismo incluir toda la información de
los estudios clínicos que sustente la combinación según los lineamientos del
Comité de Expertos de EMEA CPMP/BWP/477/97).
Los ensayos
clínicos y las informaciones que de ellos deriven deberán cumplir con las
Buenas Prácticas Clínicas internacionalmente aceptadas. En caso de ensayos
realizados en la Argentina deberán adjuntarse las autorizaciones según la Disposición ANMAT 5330/97 y sus modificatorias y el informe final del estudio aprobado por la ANMAT.
3.2.b Vacunas
convencionales
En el caso de
vacunas convencionales, y las obtenidas a partir de cepas o combinaciones de
antígenos, de eficacia y seguridad demostrada, no es imprescindible la presentación
de estudios clínicos de Fase I, II y III originales. La información referida a
eficacia deberá ser documentada sobre la base de la Bibliografía Internacional disponible. La necesidad de estudios clínicos se requerirá para la
demostración de no-inferioridad con las vacunas comercializados de eficacia y
seguridad demostrada a la fecha.
Para el caso de
vacunas convencionales que propongan nuevos esquemas de inmunización o nuevas
indicaciones de las ya autorizadas deberá presentarse información clínica que
respalde el uso solicitado.
Cuando las vacunas
a registrar sean vacunas precalificadas por la OMS, la documentación a presentar referida a la experiencia clínica será la misma que la presentada durante el
proceso de precalificación.
4.- Liberación de lote
de vacunas
Una vez inscriptas
en el REM, las vacunas serán liberadas lote a lote por la Autoridad Sanitaria. La liberación de lote es el proceso de revisión de cada lote individual
de un producto registrado, antes de aprobar su comercialización. Este proceso
se debe llevar a cabo para todas las vacunas que se comercialicen en Argentina
o a solicitud del titular del registro para su exportación. La liberación
involucra la revisión de los datos producción y de control de calidad del
fabricante por parte de la Autoridad Sanitaria y puede o no incluir la realización de ensayos de laboratorio sobre el producto terminado. La realización de
los ensayos dependerá de factores tales como: el tipo de producto, el
fabricante, el historial de producción y control del producto, la evaluación de
riesgos, el historial de comercialización del producto y la población a la que
va dirigida el producto.
Debido a que
algunas vacunas son moléculas complejas que no pueden ser químicamente
definidas y a la variabilidad característica de los sistemas biológicos, cada
proceso de producción debe ser considerado único y por esto la liberación de
lotes es indispensable en el control de vacunas.
Durante el
registro del producto, el fabricante deberá presentar para su aprobación por la
autoridad sanitaria, el proyecto de Protocolo resumido de fabricación y control
del lote. Una vez aprobado, dicho protocolo resumido será presentado por el
fabricante ante el INAME para solicitar la liberación de cada lote de vacuna.
Para obtener la
liberación de cada lote de vacuna importada, el importador deberá presentar la
siguiente documentación:
• Nombre comercial
del producto
• Principio activo
• Nombre del
producto en país de origen
• Número de
Certificado ANMAT
• Titular del
certificado en origen
• Nombre y
Domicilio del/ los fabricante/s indicando qué proceso lleva a cabo
• Nombre y
Domicilio del importador
• Número de lote
• Presentación
• Fecha de
elaboración
• Fecha de
vencimiento
• Número de
unidades a ingresar
• Fecha de ingreso
del producto a depósito
• Dirección del
Depósito habilitado en el que se encuentra el producto
• Protocolo
resumido de fabricación y control del lote
• Certificación de
liberación de lote por parte de la Autoridad Sanitaria del país de origen rubricado por escribano.
Evaluada y
aprobada la documentación aportada, el INAME procederá a emitir la liberación
del lote.
En el caso de
vacunas de producción nacional, una vez cumplido el Control de Calidad interno,
el laboratorio productor deberá presentar ante el INAME la siguiente
documentación:
• Nombre comercial
del producto
• Principio activo
• Número de
Certificado ANMAT
• Nombre y
Domicilio del/ los fabricante/s indicando qué proceso lleva a cabo
• Número de lote
• Tamaño de lote
• Presentación
• Fecha de
elaboración
• Fecha de
vencimiento
• Número de
unidades que conforman el lote
• Fecha de
aprobación por Control de Calidad
• Dirección del
Depósito habilitado en el que se encuentra el producto
• Nombre del
producto en país de origen
• Protocolo
resumido de fabricación y control del lote
Evaluada
técnicamente la documentación aportada, el INAME procederá a emitir la
liberación del lote.
• 5- Registro de
vacunas de interés sanitario en emergencias.
Para el caso de
emergencias o cuando las condiciones sanitarias hagan necesaria la
disponibilidad de vacunas en desarrollo o de reducida disponibilidad de datos
de seguridad y eficacia, la autoridad regulatoria establecerá un procedimiento
que permita evaluar las condiciones de riesgo beneficio para la disponibilidad
del biológico en el marco de la estrategia que fije el país
• 6.-
Farmacovigilancia de Vacunas
Al momento del
registro deberá presentarse un plan de vigilancia post registro
GLOSARIO
Adyuvantes:
Moléculas que incorporadas a la formulación de las vacunas aumentan la acción
inmunológica de las mismas a nivel celular y molecular.
Anticuerpos:
Proteínas producidas por Linfocitos B el organismo para defenderse en forma
específica del ataque de agentes extraños.
Buenas Prácticas
de Fabricación (BPF): Conjunto de procedimientos y prácticas destinadas a
garantizar la producción uniforme de lotes de medicamentos que satisfagan las
normas de identidad, actividad, pureza etc.
Cepas: Grupo de
organismos de una misma especie que poseen una o pocas características
distintivas en común.
Criterios de
aceptación: Especificaciones del producto y criterios de aceptación o rechazo
necesarios para tomar la decisión de aceptar o rechazar un material.
Número de lote:
Combinación distintiva de números y/o letras que identifica inequívocamente un
lote tanto en los rótulos, su registro de lote y certificados de análisis
correspondientes.
Controles en
proceso: Controles realizados durante la producción para monitorear y, si es
necesario ajustar el proceso para asegurar que el producto cumple con sus
especificaciones. El control del ambiente y equipamiento pueden también ser
contemplados como parte del control en proceso.
Diagrama de flujo:
Esquema o síntesis gráfica de todas las etapas, procesos y/o procedimientos
llevados a cabo para la obtención del producto final, detallados para cada
etapa según nivel de validación y requerimiento crítico.
Ensayos Clínico
(Estudio Clínico): Es un estudio sistemático, siguiendo todas las pautas del
método científico en seres humanos, donde participan voluntarios para estudios
con vacunas con el objeto de descubrir verificar efectos y/o identificar la
seguridad y/o inmunogenicidad y eficacia del producto en investigación.
Especificación:
Lista de requerimientos detallados con los cuales los productos o materiales
utilizados u obtenidos durante la elaboración deben cumplir. Estos sirven como
una base para la evaluación de la calidad.
Estudios de
provocación - (Estudios de desafío o reto Estudios donde se pretende demostrar
la potencia de las vacunas por exposición al agente infeccioso.
Estudios
preclínicos (Ensayos Preclínicos): Conjunto de estudios para el desarrollo de
un medicamento que se efectúan "in vitro" o en animales de
experimentación y que se diseñan para obtener la información necesaria para
decidir si se justifican estudios más amplios en seres humanos, sin exponerlos
a riesgos injustificados. Se aplican asimismo para estudiar la seguridad de
nuevos conservantes, adyuvantes.
Exposición
Sumaria: Se entiende como el resumen que sintetiza los datos más relevantes del
producto en cuanto a sus características fisicoquímicas, antecedentes de
seguridad y eficacia obtenidos en las sucesivas etapas de investigación así
como los antecedentes que demuestren la efectividad y aplicación en el contexto
donde será utilizado el producto a registrar.
Fabricación: Todas
las operaciones de transformación de materiales y productos, producción,
control de calidad, liberación, almacenamiento, transporte y distribución de
productos farmacéuticos y los controles relacionados.
Informe del
Experto: Informe técnico sobre diferentes aspectos de la forma farmacéutica,
del producto final y/o de los procedimientos finales o intermedios, o
consideraciones específicas de evaluación de ensayos técnico farmacéuticos,
preclínicos o clínicos puntuales que incluyan el punto de vista completo sobre
el tema de evaluación. Debe responder a la demostración de cumplimiento de
objetivos, por la demostración del cumplimiento de métodos y significaciones
estadísticas apropiadas con conclusiones medibles y reproducibles. Se entiende
por experto al profesional que pueda acreditar solvencia según su historia de
vida en el tema de análisis que corresponda, preferentemente independiente a
los intereses de la solicitante.
Línea (s)
celular(es): Cultivos de células que tienen alta capacidad de multiplicación
"in Vitro". En líneas celulares diploides, las células tienen la
misma característica de aquellos tejidos que la originaron. En líneas celulares
continuas las células están disponibles para multiplicarse en forma indefinida
en cultivo y pueden ser obtenidas de cultivos celulares normales o tumorales.
Algunas de las células continuas tienen potencial oncogénico bajo ciertas
condiciones
Lote: Cantidad
definida de materia prima, material de acondicionamiento o producto, elaborado
en un proceso o serie de procesos de tal manera que resulte homogéneo.
Materia prima:
Toda sustancia de calidad definida utilizada en la producción de un producto
farmacéutico.
Procedimientos
normalizados de operación (PON): Procedimiento escrito autorizado que contiene
instrucciones para llevar a cabo operaciones no necesariamente específicas para
un dado producto o material (operación de equipos, mantenimiento y limpieza,
validación, limpieza de áreas y control ambiental, muestreo e inspección).
Ciertos PON pueden ser utilizados para complementar las especificaciones
maestras del producto y la documentación de producción de lote.
Producción: Todas
las operaciones involucradas en la preparación de un producto farmacéutico
desde la recepción de los materiales, a través del procesado y
acondicionamiento hasta la obtención del producto terminado.
Producto
terminado: Forma farmacéutica final que pasó por todos los estadios de fabricación
incluyendo el acondicionamiento en el envase final.
Reproceso:
Retrabajo de todo o parte de un lote de producto de calidad inaceptable
proveniente de una etapa definida de producción, con el fin de que su calidad
pueda ser aceptada.
Vacunas: Preparaciones
conteniendo sustancias antigénicas capaces de inducir en el hombre una
inmunidad activa y específica contra un agente infeccioso, sus toxinas o
antígenos elaborados por él.
Vacunas
convencionales: Vacunas que ya cuentan con requerimientos de la Organización Mundial de la Salud, y/o monografías en Farmacopeas Internaciones y/o forman
parte de las vacunas utilizadas en los programas de inmunización como parte de
los esquemas recomendadas internacionalmente.
Vacunas Noveles
(Innovativas): Vacunas para las que no se cuenta con antecedentes de seguridad
y eficacia ya sea porque no se conoce una vacuna contra el microorganismo a
prevenir o se refiere a una nueva combinación de antígenos, nueva forma
farmacéutica, una nueva vía de administración o contiene nuevos adyuvantes o
nuevos conservadores.
Validación: Acción
documentada, en concordancia con los principios de las Buenas Prácticas de
Fabricación, que demuestra que los procedimientos, procesos, equipamientos,
materiales, actividades o sistemas conducen realmente a los resultados
previstos.

ANEXO II
INDICE DE LA PRESENTACION - TEMA / DOCUMENTO
A.- DATOS A
COMPLETAR EN EL FORMULARIO.
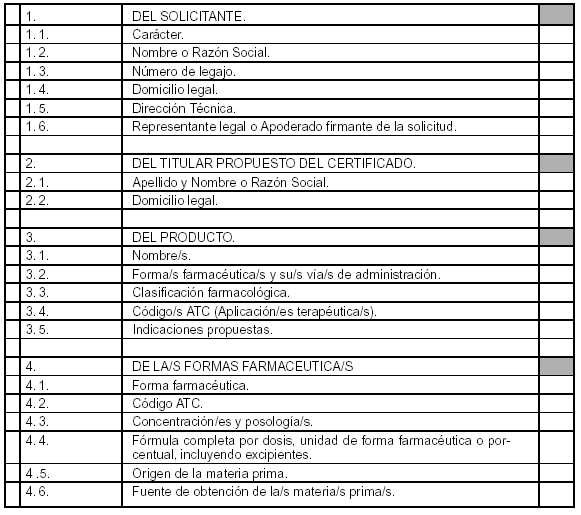
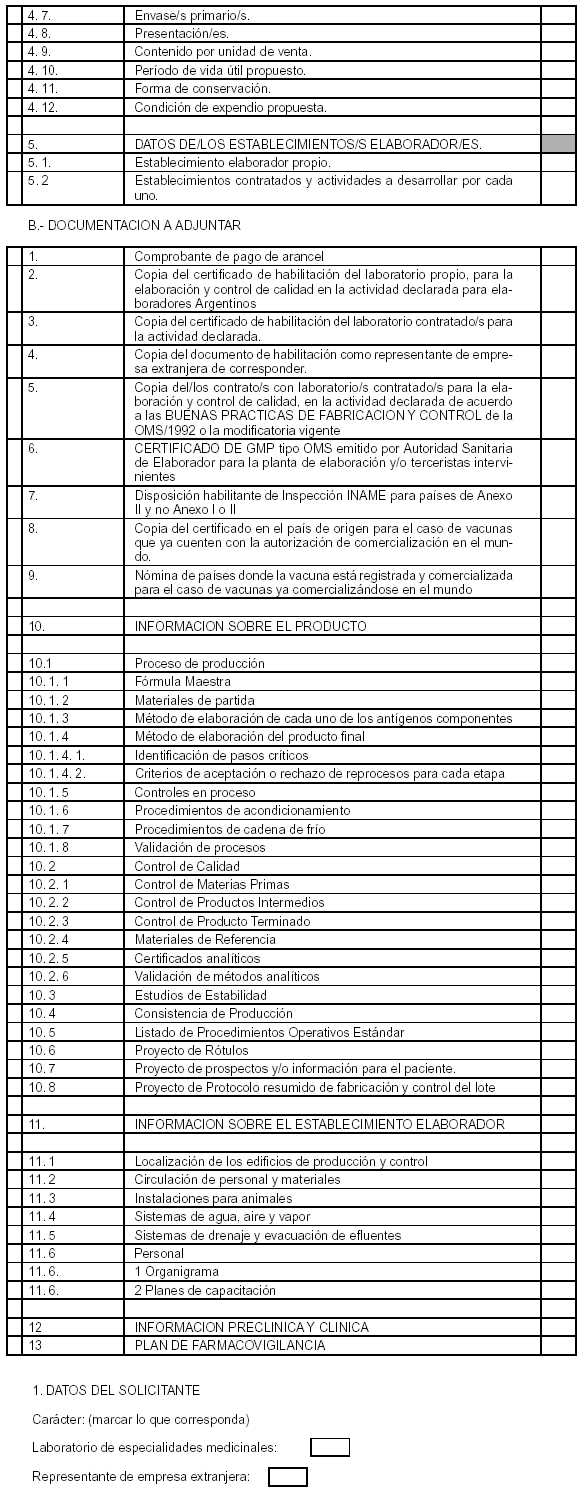



INFORMACION
CONFIDENCIAL EN LOS TERMINOS DE LA LEY 24766
DE FOJAS: A FOJAS:
ARTICULO 1º: Las
personas físicas o jurídicas podrán impedir que la información que esté
legítimamente bajo su control se divulgue a terceros o sea adquirida o
utilizada por terceros sin su consentimiento de manera contraria a los usos
comerciales honestos, mientras dicha información reúna las siguientes
condiciones:
a) Sea secreta en
el sentido que no sea, como cuerpo o en la configuración y reunión precisa de
sus componentes, generalmente conocida ni fácilmente accesible para personas
introducidas en los círculos en que normalmente se utiliza el tipo de
información en cuestión: y
b) Tenga un valor
comercial por ser secreta: y
c) Haya sido
objeto de medidas razonables, en las circunstancias, para mantenerla secreta,
tomadas por la persona que legítimamente la controla.
Se considerará que
es contrario a los usos comerciales honestos el incumplimiento de contratos, el
abuso de confianza, la instigación y adquisición de información no divulgada
por terceros que supieran o no, por negligencia grave, que la adquisición
implicaba tales prácticas.
ARTICULO 3º: Toda
persona que con motivo de su trabajo, empleo, cargo, puesto, desempeño de su
profesión o relación de negocios, tenga acceso a una información que reúna las
condiciones enumeradas en el artículo 1º y sobre cuya confidencialidad se lo
haya prevenido, deberá abstenerse de usarla y revelarla sin causa justificada o
sin consentimiento de la persona que guarda dicha información y de su usuario
autorizado.
ARTICULO 12º:
Quien incurriera en la infracción de lo dispuesto en la presente Ley en materia
de confidencialidad, quedará sujeto a la responsabilidad que correspondiera
conforme con el Código Penal y otras normas penales.