RE-52-1994
SOLUCIONES PARENTERALES DE GRAN
VOLUMEN
VISTO: El Artículo 13
del Tratado de Asunción, el Artículo 10 de Decisión No 4/91 del Consejo del
Mercado Común, la Resolución No 91/93 del Grupo Mercado Común y la Recomendación No 34/94 del SGT No 3 – “Normas Técnicas”.
CONSIDERANDO:
Que una propuesta
armonizada sobre las condiciones a cumplir en la producción y en el control de
calidades de las Soluciones Parenterales de Gran Volumen garantizará niveles
adecuados de calidad de las mismas para asegurar la preservación de la salud
pública de los Estados Partes y la lealtad de los intercambios comerciales.
Que los Estados Partes
armonicen y aprueben los contenidos del DOCUMENTO A-1/91 – SOLUCIONES
PERENTERALES DE GRAN VOLUMEN.
EL GRUPO MERCADO COMUN
RESUELVE:
Artículo 1 – Aprobar el DOCUMENTO A-1/91 – SOLUCIONES
PERENTERALES DE GRAN VOLUMEN, integrado por un REGLAMENTO TECNICO Y 11 ANEXOS
(de la A a la L), que se anexan a la presente Resolución.
Artículo 2 – Los Estados
Partes pondrán en vigencia las disposiciones legislativas, reglamentarias y
administrativas necesarias para el cumplimiento a la presente Resolución a
través de los siguientes organismos:
Argentina:
Administración
Nacional de Medicamentos y Alimentos
Brasil:
Secretaria de
Vigilancia Sanitária do Ministério da Saúde
Paraguay:
Dirección de
Vigilancia Sanitaria del Ministerio de Salud Pública y Bienestar Social
Uruguay:
Ministerio de
Salud Pública
Artículo 3 – La presente
Resolución entrará en vigencia el día 1º de enero de 1995.
XV
GMC – Brasilia, 4/XI/1994
SGT Nº 3 –
REC Nº 34/94
SOLUCIONES PARENTERALES DE GRAN
VOLUMEN
DOCUMENTO A-1/91
Versión final
Montevideo, Marzo de 1994
ANEXO
REGLAMENTO TECNICO
SOLUCIONES PARENTERALES DE GRAN
VOLUMEN
CONTENIDO
1
OBJETIVO
2
NORMAS Y
DOCUMENTOS DE REFERENCIA
3
DEFINICIÓN
4
CONDICIONES
5
CONDICIONES
ESPECÍFICAS
6
ESTERILIZACIÓN
7
PRODUCTO
TERMINADO
8
RÓTULOS
9
FECHA DE
VENCIMIENTO
10
EMPAQUE
11
DOCUMENTACIÓN
12
CONTRAMUESTRAS
13
ALMACENAMIENTO
14
TRANSPORTE
15
RECEPCIÓN,
ALMACENAMIENTO Y DISTRIBUCIÓN
16
ÍNDICE DE ANEXOS
1.
OBJETIVO
Este Reglamento Técnico (RT) estipula las condiciones que
deben cumplirse en la producción y control de calidad de soluciones
parenterales de gran volumen.
2. NORMAS Y DOCUMENTOS DE REFERENCIA
Las metodologías y especificaciones no contempladas
en el presente documento deben estar fundamentadas, en primer lugar, en la FARMACOPEA EUROPEA, en segundo lugar, en la FARMACOPEA de los EE.UU., y por último, en las
FARMACOPEAS de los países del MERCOSUR.
Las monografías contempladas en los anexos de este
documento deberán ser actualizadas cuando existan modificaciones significativas
en las Farmacopeas citadas.
3. DEFINICION
3.1. Soluciones parenterales de gran volumen.
Soluciones en base acuosa, estériles, apirogénicas , acondicionadas en
recipiente único con capacidad de 100 ml o más, esterilizadas terminalmente.
Están incluidas en esta definición las
soluciones para administración endovenosa, las soluciones para irrigación y las
soluciones para diálisis peritoneal. El término PARENTERAL DE GRAN VOLUMEN no
incluye ningún producto de origen biológico.
4. CONDICIONES GENERALES
4.1. Las empresas que intervengan en la elaboración de
los productos comprendidos en este reglamento, deben obtener autorización
previa de funcionamiento por parte de la autoridad sanitaria nacional
competente y la fabricación de dichos productos debe ser realizada bajo la
responsabilidad técnica de un profesional habilitado de acuerdo con la
legislación vigente en cada país.
4.2. En el caso de los productos importados el importador
y distribuidor son responsables legales del cumplimiento de este Reglamento.
4.3. Los establecimientos, sus equipos e instalaciones,
así como el proceso de fabricación, deben responder a las Buenas Prácticas de
Fabricación recomendadas en el Anexo A de este Reglamento.
4.4. Cada producto debe estar registrado y autorizado
en el organismo sanitario nacional competente.
5. CONDICIONES ESPECIFICAS
Para las materias primas y materiales de envase se
establecen requisitos y recomendaciones no reguladas. Los requisitos se señalan
en cada caso (RQ) en los anexos específicos.
5.1. Materias primas. El agua y las demás materias
primas utilizadas en la producción de soluciones parenterales de gran volumen
deben responder a los requisitos de calidad especificados en el Anexo B, de
este Reglamento.
5.2. Material de envase. El material de envase para
soluciones parenterales de gran volumen puede ser:
5.2.1. Vidrio. El vidrio utilizado para envases de
soluciones parenterales de gran volumen debe cumplir con los requisitos
establecidos en el Anexo C de este Reglamento.
5.2.2. Plástico. El plástico utilizado para los envases
de soluciones parenterales de gran volumen debe cumplir con los requisitos
establecidos en el Anexo D, de este Reglamento.
La utilización de cualquier plástico
no considerado en el Anexo D, en la fabricación de recipientes para Soluciones
parenterales de gran volumen, depende del cumplimiento de las exigencias de la
referida norma así como de la aprobación del material por la Autoridad Sanitaria Nacional competente y las de los países del MERCOSUR.
6. ESTERILIZACION
6.1. Proceso de esterilización. Los procesos de
esterilización de soluciones parenterales de gran volumen se deben realizar
sobre el producto envasado y cerrado empleando calor húmedo en condiciones
específicas de tiempo, temperatura y presión, de modo de asegurar una
probabilidad de sobrevida microbiana no superior a 1 x 10-6.
6.2. Validación del proceso. Los procesos de
esterilización empleados deben ser validados en forma periódica. La validación
de un proceso de esterilización debe incluir estudios de control biológico, de
distribución de temperatura y penetración de calor, de acuerdo con el Anexo H
de este Reglamento.
6.3. Control de Proceso. Para el control de proceso de
esterilización se deben realizar los siguientes procedimientos:
6.3.1. Control de temperatura con termómetro de mercurio
o su equivalente, calibrado como mínimo una vez cada 3 meses. Se deben
descartar termómetros que presenten variaciones mayores de 0,5º C en relación
al termómetro patrón.
6.3.2. Control de presión con manómetro o su equivalente
que debe ser calibrado como mínimo una vez cada 3 meses, de acuerdo con las
especificaciones del equipo.
6.3.3. Control microbiológico en cada ciclo de
esterilización utilizando indicadores microbiológicos.
6.3.4. Definición del número de lote con identificación
del equipo de esterilización y del ciclo en el cual se realiza la
esterilización. Siempre que un lote sea subdividido en esta fase, cada fracción
de lote debe ser debidamente identificada.
7. PRODUCTO TERMINADO
Los productos terminados deben ser sometidos a
controles físicos, químicos, biológicos y microbiológicos de acuerdo con los
requisitos establecidos en el Anexo E de este Reglamento.
8. ROTULOS
8.1. Requisitos generales. Los rótulos, sean adheridos
o impresos en forma indeleble sobre la superficie de los envases, deben
consignar como mínimo los siguientes datos en el idioma del país en que circule
el producto:
- Nombre del producto.
- Nombre genérico o denominación común
internacional.
- Número de registro otorgado por la Autoridad Sanitaria Nacional Competente.
- Nombre y dirección del fabricante.
- Composición cuali y cuantitativa
porcentual.
- Contenido electrolítico (mEq/L -
mmol/L).
- Osmolaridad.
- Volumen nominal.
- Número de lote.
- Fecha de vencimiento.
- Condiciones de conservación y
transporte (cuando el producto lo exija).
- Nombre del Director Técnico o
Responsable Técnico.
- Vía de administración.
- Indicaciones, contraindicaciones y
precauciones cuando sea necesario.
8.2. Tintas y colas. Las tintas usadas en los procesos
de impresión de los envases así como las colas usadas para adherir los rótulos,
no deben contener sustancias tóxicas que puedan migrar a la solución.
9. FECHA DE VENCIMIENTO
La fecha de vencimiento de las soluciones
parenterales de gran volumen debe ser determinada por estudios de estabilidad
indicados en el Anexo J del presente Reglamento y repetidos ante modificaciones
que puedan afectar la composición, calidad y/o estabilidad del producto.
10. EMPAQUE
Los materiales empleados para empacar las
soluciones parenterales de gran volumen para la venta deben ser adecuados para
proteger y mantener el producto en las condiciones normales de transporte y
traslado, de acuerdo con el Anexo A de este Reglamento.
11. DOCUMENTACION
Los documentos en que se registren los
procedimientos de producción y control de un lote deben ser archivados por el
fabricante por un período mínimo de 6 meses a partir de la fecha de vencimiento
del producto.
12. CONTRAMUESTRAS
De cada lote de fabricación se deberá conservar el
doble de las unidades requeridas para efectuar todos los análisis prescriptos,
excepto los de esterilidad y piretógenos, como mínimo hasta 30 días después de
la fecha de vencimiento, identificadas como muestras de referencia.
13. ALMACENAMIENTO
Las soluciones parenterales de gran volumen, deben
ser almacenadas en locales secos y limpios, libres de insectos y roedores y
deben cumplir con las indicaciones con respecto al estibaje de cajas y paletas,
de modo de que el almacenamiento no altere la identificación, composición y
pureza del producto, de acuerdo con lo recomendado en el Anexo A de este
Reglamento.
14. TRANSPORTE
Las soluciones parenterales de gran volumen, deben
ser transportadas de acuerdo con lo indicado en el Anexo F de este Reglamento.
15. RECEPCION, ALMACENAMIENTO Y DISTRIBUCION
La recepción, almacenamiento y distribución de SPGV
debe seguir los procedimientos descriptos en el Anexo G de este Reglamento.
16. INDICE DE ANEXOS
Anexo A: Buenas Prácticas de fabricación para SPGV
Anexo B: Materias Primas.
Anexo C: Recipientes de vidrio.
Anexo D: Recipientes plásticos para SPGV.
Anexo E: Producto Terminado.
Anexo F: Transporte de SPGV.
Anexo G: Recepción, almacenamiento y distribución
de SPGV.
Anexo H: Validación de ciclos de esterilización por
vapor.
Anexo I: Tapones de elastómero.
Anexo J: Estabilidad.
Anexo L: Ensayos biológicos
ANEXO A
BUENAS PRACTICAS DE FABRICACION
SOLUCIONES PARENTERALES DE GRAN VOLUMEN
CONTENIDO
A-1
OBJETIVO
A-2
DEFINICIONES
A-3
CONDICIONES GENERALES
A-4
INSPECCIONES
A-5 GUIA
PARA INSPECCIONES
A. 1. OBJETIVO
A.1.1. Esta recomendación fija procedimientos de Buenas
Prácticas de Fabricación a ser observado en la producción, en el control de
calidad, en el acondicionamiento y almacenamiento y en la distribución de
soluciones parenterales de gran volumen.
A. 2. DEFINICIONES
Para efecto de esta recomendación son adoptadas las
definiciones de A.2.1. a A.2.22.
A.2.1. Buenas Prácticas de Fabricación (BPF). Conjunto de
recomendaciones escritas que tienen por objeto la definición y estandarización
de procedimientos de fabricación, control de calidad, condiciones de las
instalaciones de una empresa, sus equipamientos y su respectivo mantenimiento,
también empaque y condiciones de almacenamiento de las soluciones parenterales
de gran volumen, con el objetivo de garantizar que los productos cumplan las
especificaciones establecidas con relación a su actividad, pureza, eficacia e
inocuidad.
A.2.2. Solución parenteral de gran volumen (SPGV).
Solución en base acuosa estéril y apirogénica acondicionada en recipiente único
con una capacidad de 100 mL o más esterilizada terminalmente.
Están incluidas en esta definición las
infusiones endovenosas, soluciones para irrigación y soluciones para diálisis
peritoneal. El término parenteral de gran volumen no incluye ningún producto de
origen biológico.
A.2.3. Control de Calidad (CC). Conjunto de operaciones
(programación, coordinación y ejecución) con el objetivo de verificar la
conformidad del producto con las especificaciones establecidas.
A.2.4. Fabricante. Persona jurídica que elabora SPGV, con
autorización previa de funcionamiento por parte de la autoridad sanitaria
nacional competente.
A.2.5. Fabricación. Todas las operaciones necesarias para
la obtención de los productos comprendidos en esta recomendación.
A.2.6. Lote. Cantidad de SPGV que se produce en un ciclo
de fabricación cuya característica esencial es la homogeneidad. Para el
propósito del ensayo de esterilidad, lote es el conjunto de envases preparados
de tal forma que el riesgo de contaminación pueda ser considerado el mismo para
cada unidad, normalmente es una carga del autoclave.
A.2.7. Número de lote. Designación impresa en el envase
de cada unidad de producto, constituida por combinaciones de letras, números o
símbolos, que permitan identificar el lote a que éste pertenece, y en caso de
necesidad, localizar y rever todas las operaciones de producción, inspección,
control, empaque, almacenamiento y distribución del producto en cuestión.
A.2.8. Garantía de Calidad (GC). Esfuerzo organizado y
documentado dentro de una empresa con el sentido de desarrollar, producir,
mantener y asegurar las características del producto, de modo que cada unidad del
mismo esté de acuerdo con sus especificaciones.
A.2.9. Materia Prima. Sustancia activa o inactiva que se
emplea en la fabricación de SPGV.
A.2.10. Producto semi-elaborado. Sustancia, mezcla de
sustancias o SPGV que aún se encuentren en proceso de fabricación.
A.2.11. Producto terminado. Producto con la presentación
definitiva, conteniendo ingrediente(s) activo(s) asociado(s) o no a sustancias
auxiliares, atendiendo las exigencias legales y con la aprobación final de
control de calidad del fabricante.
A.2.12. Esterilización. Proceso de aplicación de calor
húmedo al producto ya envasado y cerrado, en condiciones específicas de tiempo,
temperatura y presión, que garanticen una probabilidad de sobrevida microbiana
no superior al 1 x 10-6
A.2.13. Tiempo de esterilización. Período de tiempo, en
minutos, a una determinada temperatura y presión, durante el cual el producto
debe ser expuesto al calor húmedo, a fin de cumplir las exigencias de A. 2. 12.
A.2.14. Estudio de distribución de la temperatura. Estudio
que tiene por objeto verificar y asegurar que la temperatura en el interior del
autoclave se distribuya uniformemente durante el proceso de esterilización.
A.2.15. Estudio de penetración del calor (energía
térmica). Estudio que tiene por objeto verificar la cantidad de calor (energía
térmica) recibida por la solución durante el proceso de esterilización.
A.2.16. Cuarentena. Retención temporaria de materias
primas, productos semielaborados o productos terminados, con la prohibición de
ser utilizados hasta que sean aprobados para su uso por Control de Calidad.
A.2.17. Material de envase. Recipientes de vidrio o
plástico y tapones de elastómero que responden a las definiciones de los anexos
C, D e I.
A.2.18. Area de ambiente controlado. Area donde los factores
ambientales y de calidad del aire con relación al número de partículas y de
microorganismos son controlados.
A.2.19. Partículas extrañas. Partículas móviles
insolubles, visibles al ojo desnudo, diferentes a burbujas de gas.
A.2.20. Archivo maestro. Conjunto de documentos
conteniendo el proyecto, la formulación, las especificaciones, los
procedimientos de fabricación, los requisitos de control de calidad y los
procedimientos de acondicionamiento, de empaque y de almacenamiento del
producto.
A.2.21. Archivo histórico. Conjunto de documentos
conteniendo los registros de producción y control de cada lote de fabricación,
en forma ordenada, archivado bajo la responsabilidad de CC.
A.2.22. Filtro Hepa. Filtro para aire de alta eficiencia
con la capacidad de retener 99,97% de las partículas mayores de 0,3 µm de
diámetro.
A.3. CONDICIONES GENERALES.
A.3.1. Organización y personal.
A.3.1.1. Estructura orgánica y de personal.
A.3.1.1.1. Todo fabricante de SPGV debe poseer
una estructura orgánica y de personal suficiente para garantizar que los
productos por él fabricados estén de acuerdo con los requisitos de esta
recomendación.
A.3.1.1.2. Toda empresa productora de SPGV debe
poseer un sistema de control de calidad que ejerza sus actividades plenamente,
de modo de garantizar la calidad de los productos fabricados.
A.3.1.1.3. Todo fabricante debe adoptar e
implementar procedimientos referidos a la calidad, formalmente establecidos y
documentados y que hayan sido aprobados específicamente para el producto a ser
fabricado.
A.3.1.1.4. El responsable legal de los productos
fabricados será un Farmacéutico. Los responsables del control de calidad y de
la producción deben ser profesionales calificados, con conocimientos en las
áreas de química, físico-química, bioquímica, microbiología, farmacología,
farmacotecnia, tecnología farmacéutica y toxicología. Deben también poseer
experiencia práctica en los procesos de producción y de control de calidad,
para el cumplimiento de sus atribuciones con discernimiento e independencia
profesional.
A.3.1.2. Responsabilidad de Control de Calidad. El
responsable de CC tiene autoridad para:
a) aprobar o rechazar todas las materias primas,
materiales auxiliares de fabricación, materiales de empaque y rótulos, como así
también los productos semi-elaborados y terminados;
b) rever y revisar, en cualquier momento, los
registros de producción, a fin de asegurar que no fueron cometidos errores y,
si éstos ocurrieron, que hayan sido debidamente corregidos e investigadas sus
causas;
c) acompañar cada proceso de fabricación para
certificar que los métodos de producción preconizados están siendo seguidos,
así como verificar si los límites de seguridad, en cada etapa de fabricación,
están de acuerdo a las especificaciones;
d) identificar, recomendar o presentar soluciones
para los problemas de la calidad, así como supervisar e implementar dichas
soluciones;
e) verificar los procedimientos utilizados en las
inspecciones, con relación a su adecuación y ejecución;
f) realizar estudios de estabilidad física, química y
biológica del producto terminado, a fin de establecer su fecha de vencimiento;
g) fiscalizar técnicamente la aplicación de esta
norma en los posibles contratos de fabricación con terceros.
A.3.1.3. Requisitos de personal, entrenamiento e higiene.
A.3.1.3.1. El fabricante debe disponer de
personal calificado y en cantidad suficiente para que todas las operaciones
sean realizadas correctamente.
A.3.1.3.2. Todo el personal implicado en la
fabricación, procesamiento, empaque, transporte interno, almacenamiento y CC,
debe recibir instrucciones y entrenamiento para el perfecto desempeño de sus
funciones específicas. Periódicamente deben cumplirse programas de
entrenamiento y reciclaje que proporcionen al personal el entendimiento
completo de sus actividades y la importancia del cumplimiento de las BPF. La
ejecución de tales programas debe ser documentada. La realización de esos
programas debe ser siempre en forma continua y documentada, y debe darse
conciencia al personal de la importancia del cumplimiento de las BPF.
A.3.1.3.3. Los responsables de la Calidad deben estar informados de todos los inconvenientes que pueden ser encontrados en la
ejecución de cada operación de producción y de control.
A.3.1.3.4. Todo personal responsable de la
supervisión de producción, procesamiento, empaque, transporte interno,
almacenamiento y CC, debe recibir instrucciones y entrenamiento, y tener
experiencia para garantizar que el producto cumpla las especificaciones de
identidad, pureza y concentración.
A.3.1.3.5. Debe haber un número adecuado de
personal calificado para desempeñar y supervisar todas las etapas mencionadas
anteriormente.
A.3.1.3.6. El personal en contacto con el
producto o el ambiente de fabricación, debe presentar condiciones de salud e
higiene de modo de evitar contaminación del producto; ese personal debe ser
sometido periódicamente a exámenes médicos y en caso de que haya portadores de
enfermedades infecto-contagiosas, estos deben ser desafectados temporaria o
definitivamente de sus actividades. El personal debe ser instruido para referir
a sus supervisores cualquier alteración en su estado de salud.
A.3.1.3.7. El personal implicado en la
producción y CC tendrá que disponer de uniformes que deben ser cambiados para
garantizar la higiene apropiada.
A.3.1.3.8. Las vestimentas deberán adaptarse al
proceso y a los lugares de trabajo, de tal manera que aseguren la protección
del producto frente a la contaminación.
A.3.1.3.9. La colocación de los uniformes y el
calzado, así como la higiene previa a la entrada en las áreas de llenado y
control microbiológico, deben ser realizadas en lugares específicamente
designados para vestuario, de acuerdo con las exigencias estipuladas en A.
3.2.2.
A.3.1.3.10. El acceso a las áreas de ambiente
controlado debe ser limitado a personas debidamente entrenadas, de modo de
mantener la integridad ambiental.
A.3.1.3.11. El personal de empaque y de
transporte interno, así como los auxiliares indirectos (mantenimiento), deben
usar uniformes igualmente limpios.
A.3.1.3.12. Cualquier persona que evidencie una
condición inadecuada de higiene o una vestimenta que pueda afectar al producto,
debe ser desafectada de sus actividades hasta que tal condición sea corregida.
A.3.2. Edificaciones y control ambiental.
A.3.2.1. Areas. Características generales.
A.3.2.1.1. Las áreas de fabricación de SPGV
deben tener dimensiones adecuadas para facilitar al máximo la limpieza, el
mantenimiento y las operaciones de procesamiento.
A.3.2.1.2. Todas las áreas involucradas directa
o indirectamente en la fabricación de SPGV deben presentar pisos, paredes y
revestimientos lisos, impermeables y fácilmente lavables. En las áreas donde
hay ventanas, éstas deben ser protegidas con telas para evitar la entrada de
insectos, aves y roedores.
A.3.2.1.3. Cada área debe tener espacio
suficiente para la colocación ordenada del equipamiento y los materiales. A fin
de permitir un flujo racional de trabajo y manejo seguro de las operaciones
relacionadas con el ítem A.3.2.1.5.
A.3.2.1.4. Cada área debe ser perfectamente
individualizada para que sea mínimo el riesgo de contaminación cruzada.
A.3.2.1.5. Las operaciones deben ser realizadas
dentro de áreas definidas específicamente y de dimensiones adecuadas para:
a) recepción de materias primas, material de
acondicionamiento y rótulos;
b) cuarentena (materias primas, material de
acondicionamiento y rótulos);
c) almacenaje de materias primas, materiales de
acondicionamiento y rótulos aprobados;
d) almacenaje de materias primas, materiales de
acondicionamiento y rótulos rechazados hasta su devolución o destrucción;
e) producción propiamente dicha;
f) llenado;
g) esterilización;
h) rotulado y empaque;
i) cuarentena de productos en proceso;
j) almacenaje de productos terminados - expedición;
k) control de calidad.
A.3.2.1.6. Actividades tales como alimentación,
fumar y recreación deben ser restringidas a áreas aisladas, limitadas y
determinadas.
A.3.2.2. Areas de ambiente controlado. En las áreas
de ambiente controlado, las paredes, pisos, techos, accesorios y divisores
deben tener superficies lisas e impermeables para permitir la limpieza
rigurosa. Deben ser construidas con materiales que resistan las rajaduras,
chispas, oxidación u otro tipo de deterioro. Estas áreas deben tener
temperatura y humedad controladas, suministro de aire filtrado con presión
positiva, con filtro de eficacia adecuada, sistema de limpieza y desinfección
de las salas y los equipos, cañerías de agua y aire comprimido, así como los
conductos eléctricos identificados e instalados de modo de evitar al máximo,
cualquier cúmulo de impurezas. Deben ser previstas áreas específicamente
destinadas para la colocación de vestimentas y calzados para la entrada a las
áreas de ambiente controlado. Sobre la línea de llenado y operaciones de
control microbiológico, se requerirá un ambiente con no más de 3,5 partículas
de 0,5 µm o mayores por cada litro de aire (área clase 100). En las áreas destinadas
al pesado y mezclado, se requerirá un ambiente de no más de 3.500 partículas de
0,5 µm por cada litro de aire (área clase 100.000).
A.3.2.3. Reservorios de agua potable. Los
reservorios de agua potable deben ser debidamente protegidos, a fin de evitar
contaminaciones, sea por microorganismos, insectos o aves; deben ser
construidos con materiales adecuados e impermeabilizados para evitar
infiltraciones, facilitar limpiezas periódicas e inspecciones.
A.3.2.4. Control ambiental. Debe realizarse un
riguroso control ambiental para que se evite la contaminación de los productos
y sean satisfechas las condiciones exigidas para la ejecución de las
operaciones mencionadas en A.3.2.1.4.
El protocolo para el control ambiental
debe contener como mínimo las siguientes variables: presión y filtración de
aire, aireación, temperatura, humedad, contaminación de aire, de superficies y
de las áreas de trabajo.
A.3.2.5. Iluminación. Las instalaciones de iluminación
artificial, en las áreas de producción y envasamiento, deben ser construidas de
modo de prevenir, cualquier cúmulo de polvo y facilitar la limpieza. También
deben ser protegidas para evitar rotura y dispersión de fragmentos.
A.3.2.6. Limpieza.
A.3.2.6.1. Todas las áreas de producción
mencionadas en el ítem A.3.2.1., inmediatamente después del término de cada
jornada de trabajo, deben limpiarse con agua y jabón, desinfectadas, mantenidas
limpias, en condiciones sanitarias adecuadas y estar libres de infestación por
roedores, pájaros e insectos.
A.3.2.6.2. Los procedimientos de limpieza e
higienización , también como el empleo de raticidas, insecticidas, fungicidas y
desinfectantes, deben ser cumplidos estrictamente. El uso de esos agentes debe
ser registrado por escrito y firmado por el responsable de la operación.
A.3.2.6.3. Detritus y desechos industriales,
tales como agentes químicos, residuos mecánicos u otros, eventualmente
resultantes del procesamiento del producto, deben ser eliminados en forma
segura para evitar contaminación ambiental.
A.3.2.7. Area de esterilización. Las áreas de
esterilización deben ser diseñadas y equipadas adecuadamente, o en su defecto,
adoptarse sistemas de codificación convenientes, para evitar la confusión entre
materiales y productos esterilizados o no esterilizados.
A. 3.3. Equipamiento.
A. 3.3.1. Localización e instalación. Todo
equipo utilizado en el proceso de fabricación debe ser localizado e instalado
de modo de facilitar su operación, mantenimiento, ajuste, calibración/ aforado
y limpieza. Todos los equipos de producción, incluidos los de medición,
utilizados en la producción o en los ensayos de los productos, que sean
mecánicos, neumáticos o manuales, deben ser apropiados para sus propósitos y
capaces de reproducir resultados válidos, y deben ser inspeccionados
rutinariamente, aforados y/o calibrados siguiendo procedimientos y
especificaciones escritas y debidamente registradas.
A.3.3.2. Procedimiento de limpieza.
A.3.3.2.1. Deben existir y estar disponibles
para el personal responsable, todos los cronogramas y procedimientos escritos
de limpieza, conforme a los requisitos del proceso.
A.3.3.2.2. Deben existir procedimientos escritos
para evitar la contaminación de las instalaciones, de los equipos de ensayo y
de producción, de los componentes y material y de los productos terminados, por
el uso de sustancias tóxicas de limpieza, de desinfección o de productos
químicos volátiles y corrosivos.
A.3.3.3. Procedimiento de operación. Deben
existir procedimientos escritos que orienten la operación de los equipos y que
estén fácilmente disponibles para los operadores.
A.3.3.4. Mantenimiento. Todos los equipos deben
ser sometidos a mantenimiento preventivo o correctivo, de acuerdo a los
respectivos manuales de fabricación. Los procedimientos realizados deben
registrarse por escrito.
A.3.3.5. Inspección.
A.3.3.5.1. Inspecciones destinadas a la
verificación de los procedimientos de mantenimiento y limpieza, deben ser
realizadas periódicamente y registradas de modo adecuado.
A.3.3.6. Calibración y aforado.
A.3.3.6.1. Los procedimientos de calibración y
aforado deben incluir instrucciones específicas, así como explicar los errores
tolerables. Las instrucciones para las acciones correctivas deben ser
claramente indicadas dentro de los límites tolerados.
La calibración y aforado solo deben
ser ejecutadas por personal entrenado y capacidad para operar con el equipo.
Las especificaciones y aforados deben ser recomendadas por el órgano oficial
del país. Si no existen especificaciones para algún parámetro particular, puede
ser utilizada una aceptada internacionalmente.
A.3.3.6.2. Tolerancias o limitaciones inherentes
al protocolo de operación de un equipo, deben ser fijadas sobre el mismo o
mantenidas fácilmente disponibles para el personal que lo opera o calibra.
A.3.3.6.3. Registro de calibración y aforado.
Calibraciones y aforados deben ser ejecutados de acuerdo con los procedimientos
establecidos y los resultados deben ser archivados con el nombre del
responsable. Las etiquetas con datos referidos a la última y a la próxima
calibración/ aforado deben ser adheridas a cada equipo.
A.3.3.7. Filtros. Los filtros utilizados en la
producción de SPGV no deben liberar fibras. Cuando estos filtros fueran
necesarios en el proceso, debe existir un procedimiento adicional que retenga
tales fibras. Se debe controlar la integridad física de los filtros de membrana
utilizados para la filtración final de las SPGV.
A.3.3.8. Autoclave.
A.3.3.8.1. Un autoclave para la esterilización
de SPGV debe estar equipado, como mínimo, con termómetros de mercurio o
equivalentes, adecuados para el rango de temperatura en el que se pretende
trabajar y cuya resolución máxima sea de 1 ºC. El autoclave debe poseer además, manómetro, registrador de temperatura, y válvulas de: entrada y salida de
vapor, salida de aire y de seguridad.
A.3.3.8.2. El fabricante de SPGV debe validar
cada autoclave, según lo establecido en el Anexo H..
A.3.3.9. Equipamiento auxiliar. Todos los
equipos auxiliares que abastezcan aire, agua de limpieza o agua para
fabricación, deben cumplir las recomendaciones del ítem A.3.4. de esta
recomendación.
A.3.4. Calidad de aire y agua.
A.3.4.1. Requisitos generales. Las instalaciones y
procedimientos utilizados en el procesamiento y distribución deben ser
aprobados por CC. Los resultados de todos los ensayos deben ser registrados y
mantenidos a disposición del personal responsable del proceso.
A.3.4.2. Aire en ambientes controlados.
A.3.4.2.1. El aire en ambientes controlados debe
tener un sistema de filtración que asegure:
a) para áreas clase 100.000, recuentos automáticos
máximos de 3.500 partículas de 0,5 µm o mayores por cada litro de aire o
recuentos microscópicos máximos de 25 partículas de 5 µm o mayores por cada
litro de aire;
b) para áreas clase 100, recuentos automáticos con un
máximo de 3,5 partículas de 0,5 µm o mayores por litro de aire. En las áreas de
ambiente controlado debe además mantenerse la temperatura entre 19 ºC y 25ºC, humedad relativa entre 30% y 50%, presión positiva diferencial de 0,127 cm de columna de agua, con todas las puertas cerradas, con relación a los ambientes adyacentes
menos limpios y como mínimo, 20 cambios de aire/hora. Estas especificaciones
pueden ser restringidas al área de un equipamiento o local donde sean
necesarias tales condiciones de aire.
A.3.4.2.2. El aire comprimido debe ser exento de
agua, aceite y vapores de aceite e hidrocarburos, sufriendo filtraciones
suficientes para que se obtenga, como máximo 3,5 partículas de 0,5 µm o mayores
por litro, cuando se lo utiliza en líneas de llenado, en autoclaves, en
ambientes controlados y en áreas de ensayos microbiológicos, a menos que la
descarga se realice en áreas de ambiente no controlado.
A.3.4.3. Agua
A.3.4.3.1. El agua de limpieza o lavado inicial
de superficies que van a estar en contacto con el producto tal como envases,
tapones y equipos, puede tener un máximo de 50 microorganismos por cada 100 mL,
en tres muestras consecutivas de 250 mL tomadas en el mismo punto de muestreo.
Si el agua contiene agentes bactericidas estos deberán ser neutralizados.
A.3.4.3.2. El agua para fabricación de las SPGV
y para el enjuague final de equipamientos o superficies en contacto con el
producto debe responder a las especificaciones del Anexo B "Aguas para
inyectables".
A.3.4.3.3. Cuando el agua para la fabricación
necesita de almacenamiento, deben ser usados recipientes de acero inoxidable
sanitario, herméticos y munidos de filtros de venteo de 0,22 µm de tamaño de
poro, a una temperatura no menor de 80º C, bajo constante circulación.
A. 3.4.3.4. El agua utilizada para la
refrigeración del producto después de esterilizado debe ser filtrada
bacteriológicamente para mantener la calidad del producto.
A.3.4.4. Control del aire y del agua. Los procedimientos de
control del aire y del agua deben ser escritos y aprobados por CC y la
inspección debe ser programada periódicamente. Las alteraciones y correcciones
deben ser documentadas. Todas esos documentos deben ser mantenidos a
disposición de los responsables de la producción.
A.3.4.4.1. El aire comprimido utilizado en áreas
de ambiente no controlado puede presentar como máximo 3500 partículas de 0,5 µm,
o mayores, por litro de aire.
A.3.5. Control de materias primas y de materiales de
acondicionamiento.
A.3.5.1. Exigencias generales.
A.3.5.1.1. Los procedimientos con relación a la
recepción, identificación, almacenamiento, manipulación, muestreo, ensayos y
aprobación o rechazo de materias primas y materiales de acondicionamiento deben
ser detalladamente descriptos.
A.3.5.1.2. Las materias primas y materiales de
acondicionamiento deben ser manipulados y almacenados de modo de evitar la
contaminación.
A.3.5.1.3. Materias primas, embolsadas o
acondicionadas en cajas, deben ser almacenadas de modo de no entrar en contacto
con el piso y adecuadamente posicionadas de modo de permitir limpieza e inspecciones.
A.3.5.1.4. Cada lote de materia prima y material
de acondicionamiento debe ser identificado con un código distinto siendo
apropiadamente identificado en cuanto a su situación (por ejemplo, cuarentena,
aprobado o rechazado).
A.3.5.2. Recepción y almacenamiento.
A.3.5.2.1. En la recepción de materias primas y
materiales de acondicionamiento cada lote debe ser examinado en cuanto a
rótulos, contenido y a posibles daños en su embalaje.
A.3.5.2.2. Cada lote de materia prima y material
de acondicionamiento debe ser mantenido fuera de uso hasta ser aprobado por CC.
A.3.5.3. Muestreo y ensayos para la aprobación o el
rechazo.
A.3.5.3.1. Muestras representativas de cada lote
deben ser tomadas para el análisis. El número de unidades a ser muestreado y la
cantidad de material a ser tomada de cada lote debe basarse en criterios
estadísticos para la variabilidad, niveles de confianza y grado de precisión
deseados de acuerdo con la calidad o la historia del proveedor.
A.3.5.3.2. La cantidad de muestra recogida debe
ser suficiente para la realización de todos los análisis y se debe conservar
hasta 30 días después del vencimiento del último lote con ella producida.
A.3.5.3.3. Las muestras de materias primas deben
ser recogidas de acuerdo a los siguientes procedimientos:
a) Los embalajes del material seleccionado deben ser
limpiados externamente;
b) Los embalajes deben ser abiertos, tomadas las
muestras y luego cerrados para evitar la contaminación de sus contenidos;
c) Equipamiento estéril y técnicas de muestreo
asépticas deben ser utilizadas cuando es necesario;
d) Es necesario proceder al muestreo del material
localizado en la parte superior, media e inferior de un envase y estas muestras
no pueden ser mezcladas para el ensayo;
e) Las muestras deben ser identificadas con la
información del nombre del material muestreado, el número del lote, el envase
del cual la muestra fue tomada, la fecha de la toma y el nombre de la persona
que la colectó;
f) Los envases, de los cuales fueron tomadas las
muestras, deben ser marcados para identificar su origen.
A.3.5.3.4. Las muestras de materias primas deben
ser analizadas por el fabricante de SPGV de acuerdo a lo especificado en el
anexo B. Los recipientes y tapas deben ser analizados de acuerdo a lo
establecido en los anexos C, D e I.
A.3.5.3.5. Cualquier lote de materia prima,
material de acondicionamiento que esté de acuerdo con las especificaciones
exigidas y conforme a lo previsto en el ítem 3.5.3.4. puede ser aprobado y
liberado para su uso. Caso contrario debe ser rechazado.
A.3.5.3.6. Periódicamente se deben practicar
ensayos microbiológicos, de acuerdo con un programa escrito, sobre muestras
representativas de todas las materias primas y materiales de acondicionamiento,
incluso aquellas que no se consideren susceptibles de contaminación microbiana.
A.3.5.3.7. Periódicamente se deben practicar
ensayos de sustancias piretógenas sobres muestras representativas de materias
primas y material de envase.
A.3.6. Control de producción y proceso.
A.3.6.1. Procedimientos escritos.
A.3.6.1.1. El fabricante de SPGV debe tener por
escrito los procedimientos de producción y control de sus productos.
A. 3.6.2. Materia prima.
A.3.6.2.1. Los procedimientos escritos de
control y de producción deben asegurar que el producto tenga identidad,
concentración, pureza y otros requisitos especificados, y deben incluir los
siguientes datos:
a) el lote del producto debe ser formulado con la
intención de suministrar como mínimo el 100% de la cantidad del componente
activo declarado en el rótulo;
b) las materias primas deben ser fraccionadas,
pesadas o medidas apropiadamente. Si a una materia prima le fue cambiado el
envase original por otro, el nuevo envase debe ser identificado con las
siguientes informaciones:
- nombre de la materia prima;
- número de lote;
- fecha de recepción y/o número del
control;
- peso o medida del nuevo envase;
c) la medida u operación de fraccionamiento para las
materias primas debe ser supervisada. Cada envase de materia prima preparada
para la fabricación debe ser examinado por una segunda persona para asegurar
que:
- la materia prima fue liberada por
CC.
- la medida es correcta, como consta
en los registros de producción del lote;
- los envases están adecuadamente
identificados.
A.3.6.3. Cálculo de rendimiento en la fabricación. Las
pérdidas que ocurran en la fabricación de cada lote, unidades y pérdida de
volumen de solución en cañerías y equipos deben ser identificadas, estimadas en
cantidad y registradas.
A.3.6.4. Identificación del equipamiento.
A.3.6.4.1. Todos los equipos, tanques, filtros,
bombas y cañerías utilizados durante la producción de un lote deben ser
identificados durante todo el tiempo para indicar sus contenidos y cuando sea
necesario, la etapa de fabricación. Tal identificación debe ser incorporada a
la documentación de la producción del referido lote.
A.3.6.4.2. Todos los cestos del esterilizador,
vagones, carros o cualquier otro dispositivo utilizado para retener el producto
durante el proceso de esterilización, deben estar marcados con el número del
lote en forma visible y clara, a menos que éste figure previamente impreso en
forma indeleble en cada envase.
Se deberá identificar si fueron
expuestos (los cestos) o no al proceso de esterilización.
A.3.6.5. Muestreo de materiales en proceso y producto
terminado.
A.3.6.5.1. Para asegurar la uniformidad e
integridad de un producto, deben ser establecidos procedimientos y programas
escritos para el muestreo y análisis estadísticamente válidos a fin de que
estos incluyan los siguientes aspectos:
a) conformidad de la solución antes del envasado;
b) nivel de envasamiento o volumen medio (contenido
líquido)
c) limpidez de la solución
d) ausencia de partículas
e) vacío, donde se aplique, u otros índices de
impermeabilidad del cierre
f) identidad del producto
g) caracterización del producto
h) concentración de materia prima en el producto
i) pH
j) recuento microbiano
k) piretógenos.
A.3.6.5.2. Los materiales en proceso deben ser
analizados para determinar identidad, concentración, pureza y ser aprobados o
rechazados por CC durante la producción, en el inicio o al final de las fases
significativas o después del almacenamiento por períodos prolongados.
A.3.6.6. Limitaciones de tiempo en la
producción. El tiempo que transcurre entre la adición de la primera materia
prima del producto al agua en el tanque de mezcla y la exposición de la última
unidad envasada al inicio de la esterilización no debe exceder las 8 horas.
Cuando sea necesario, deben ser
establecidos límites de tiempo para la conclusión de cada fase de la
producción, a fin de asegurar la calidad del producto. Modificaciones en el
tiempo establecido, deben ser validadas y documentadas de modo que no haya
compromiso de la calidad del producto.
A.3.6.7. Filtración final.
A.3.6.7.1. Antes del envasado, las SPGV deben
ser filtradas a través de filtros con porosidad no superior a 0,45 µm,
modificaciones en la porosidad de los filtros deberán ser validadas y
documentadas de modo que no exista compromiso en la calidad del producto.
A.3.6.7.2. La filtración final de las soluciones
debe ser realizada inmediatamente antes de su envasamiento. Las
especificaciones del proceso deben en forma adecuadamente documentada indicar
el tiempo máximo durante el cual el sistema de filtración debe ser usado, de
modo de impedir el desarrollo de microorganismos a niveles que puedan afectar
la calidad del producto. Se recomienda el reemplazo o la limpieza de los
filtros en períodos no mayores a 8 horas.
A.3.6.7.3. Los filtros deben ser ensayados de
acuerdo a un procedimiento escrito para verificar su integridad.
A.3.6.8. Inspección visual. Todas las SPGV
deben ser examinadas visualmente contra fondos oscuros y claros luego de la
esterilización, para verificar la presencia o no de partículas , la fuente y
dirección de la luz debe intensificar la visibilidad de las partículas. Un
método alternativo de examen puede ser empleado, mientras sea más preciso que
el examen visual. Se deben mantener registros del método ejecutado y de los
resultados encontrados.
A.3.6.9. Control de la contaminación
microbiana. Procedimientos escritos, destinados a evitar la contaminación
microbiana de productos que deben ser estériles, serán establecidos y seguidos.
A.3.7. Esterilización.
A.3.7.1. Proceso de esterilización. El proceso de
esterilización de las SPGV debe emplear calor húmedo realizándose sobre el
producto envasado y cerrado, en condiciones específicas de tiempo, temperatura
y presión, de modo de asegurar una probabilidad de sobrevida microbiana no superior
a 1 x 10-6, diseñado para ser reproducible, uniforme y eficiente. El
proceso de esterilización empleado debe ser validado por CC. Los registros de
validaciones deberán quedar archivados en el archivo maestro para referencia
futura. Los registros de las condiciones de esterilización obtenidos en cada
ciclo de esterilización deben ser revisados por CC y archivados junto con los
demás documentos del lote en el archivo histórico.
A.3.7.2. Validación del proceso de esterilización.
A.3.7.2.1. La validación del proceso de
esterilización debe realizarse en forma periódica. La validación de un proceso
de esterilización debe incluir estudios de control biológico, de distribución
de temperatura y penetración de calor de acuerdo con lo establecido en el Anexo
H.
A.3.7.3. Control del proceso de esterilización.
Para el control del proceso de esterilización los siguientes procedimientos
deben ser observados:
a) control de temperatura con termómetro de mercurio
o equivalente, calibrado/aforados contra un termómetro patrón, como mínimo una
vez cada 3 meses. Si hubieran variaciones mayores de símbolo ± 0,5ºC, el
termómetro no debe ser utilizado;
b) control de presión con manómetro o equivalente,
que debe ser calibrado/aforado no menos de una vez cada 3 meses, de acuerdo con
las especificaciones del equipo.
c) definición del número de lote o control
suficientemente claro para identificar al autoclave y al ciclo en el cual
ocurre la esterilización, siempre que un lote sea subdividido en esta fase;
d) revisión por CC de los registros de las
condiciones de esterilización obtenidas en cada ciclo y archivo de los mismos
junto con los demás documentos del lote en el archivo histórico;
e) el tiempo de exposición del proceso
desesterilización se comienza a contar solamente cuando la temperatura de
esterilización se alcanza tanto en la cámara como en el interior de la
solución;
f) si los registradores de temperatura mostraran
diferencias superiores a 0,5 ºC con relación a los termómetros, se vuelve
necesaria la verificación de los mismos luego de la finalización del ciclo de
esterilización donde fue detectada la diferencia;
g) todos los procedimientos, alteraciones,
calibraciones, ensayos o reposiciones verificados en los ítems a) y b) deben
ser documentados;
h) control microbiológico en cada ciclo de
esterilización utilizando indicadores microbiológicos.
A.3.8. Control de reprocesamiento. Procesamientos escritos
deben ser establecidos y seguidos, para el reprocesamiento de partidas de SPGV
que se presenten en desacuerdo con los patrones o especificaciones. Deben ser
tomadas medidas para asegurar que las partidas reprocesadas se presenten de
acuerdo con las especificaciones establecidas. El reprocesamiento no debe ser
ejecutado sin la revisión y autorización de CC.
A.3.9. Control de rotulado y empaque.
A.3.9.1. Deben existir procedimientos escritos, a ser
seguidos, describiendo en forma detallada, la recepción, identificación,
almacenamiento, manipulación, muestreo y ensayo de materiales de rotulado y
acondicionamiento. Los materiales de rotulado y acondicionamiento deben tener
muestras representativas a ser analizadas por CC antes de su uso; sólo pueden
ser liberados los materiales de acondicionamiento y rotulado que cumplan con
las especificaciones aprobadas.
A.3.9.2. Los materiales empleados para empacar
las SPGV, deben proteger y mantener el producto inalterado en las condiciones
usuales de expedición y manipulación.
A.3.10. Almacenamiento y distribución:
A.3.10.1. Procedimientos escritos orientando el
almacenamiento de las SPGV deben incluir directivas específicas, con relación
al apilamiento de cajas o palets, de forma que el almacenamiento no dañe el
producto.
A.3.10.2. Procedimiento de distribución. Deben
ser establecidos y seguidos procedimientos escritos sobre los cuidados que se
deben tener en la manipulación de una SPGV durante su distribución.
A.3.11. Documentación.
A.3.11.1. Principios y requisitos generales.
A.3.11.1.1. El sistema de documentación debe
incluir cada lote de producto, incluyendo la utilización y provisión de cada
materia prima, materiales de acondicionamiento, productos semielaborados y
productos terminados, para asegurar que el personal de producción y CC hayan
recibido instrucciones detalladas y adecuadas, relativas a los procedimientos,
y por ende permitir la investigación y seguimiento de los productos fabricados.
A.3.11.1.2. Para facilitar y efectivizar su uso,
los documentos deben ser diseñados y preparados con cuidado, destacando los
siguientes puntos:
a) el título debe ser objetivo y el documento debe
exponer su contenido con claridad para evitar interpretaciones ambiguas;
b) el flujo de circulación de los documentos debe ser
definido;
c) el tamaño, la forma, la calidad y la coloración del
papel de los documentos deben ser considerados en relación con su facilidad de
manipulación y reproducción;
d) Los documentos reproducidos deben ser claros;
e) Los documentos deben ser preparados, fechados y
firmados por un técnico y ratificados, fechados y firmados por otro legalmente
responsable;
f) los documentos deben ser periódicamente revisados,
las alteraciones deben ser hechas de tal manera que el original no sea
destruido y que las correcciones sean firmadas y fechadas;
g) el período de conservación de los documentos debe
ser:
- en el caso del archivo maestro:
permanente, al estar constituido por documentos originales, acrecentados por
revisiones efectuadas;
- en el caso del archivo histórico:
deben ser mantenidos hasta 6 meses luego del vencimiento del plazo de validez
del lote del producto.
A.3.11.2. Archivo maestro. El archivo maestro
debe existir para cada producto a ser manufacturado. A fin de asegurar
uniformidad de los lotes, de los procesos de producción y de los registros de
control. El archivo maestro debe constar de:
A.3.11.2.1. Todos los documentos iniciales que
generaron el producto.
A.3.11.2.2. Información técnica.
a) nombre del producto, composición y descripción de
la forma farmacéutica;
b) nombre y cantidad de cada materia prima y material
de acondicionamiento.
A.3.11.2.3. Especificaciones de materias primas y
de materiales de acondicionamiento, declarando:
a) nombre, descripción;
b) instrucciones de muestreo;
c) ensayos de identificación y pureza,
características químicas, físicas y biológicas;
d) métodos de ensayo utilizados;
e) catastro de proveedores;
f) precauciones a ser observadas;
g) condiciones de almacenamiento;
h) procedimientos para el ensayo del material
almacenado;
A.3.11.2.4. Procedimiento de fabricación
conteniendo:
a) local de fabricación;
b) equipamiento a ser utilizado;
c) métodos a ser utilizados para la preparación de
los equipos (limpieza, calibración, esterilización y otros);
d) etapas detalladas de fabricación:
- controles de materias primas usadas;
- pretratamiento de materias primas
cuando sea necesario;
- secuencia del agregado de las
materias primas;
- definición del sistema de
filtración;
- tiempo de mezclado y esterilización;
- temperaturas en las diversas etapas;
- cuidados especiales;
e) descripción de los recipientes para el
acondicionamiento del producto, cierres y materiales de empaque, incluyendo un
ejemplar o copia de cada rótulo y todos los otros rótulos, firmados y fechados
por las personas responsables y/o designadas para tal fin;
f) rendimiento teórico y desvíos permitidos, para
establecer la necesidad o no de investigación;
g) métodos de control en proceso con instrucciones de
muestreo y límites de aceptación;
h) criterios de aprobación para liberación del lote.
A.3.11.2.5. Especificaciones y métodos de ensayo
para liberación de producto terminado.
A.3.11.2.6. Informe de las alteraciones que
involucraron al producto desde la primer producción.
A.3.11.2.7. Otros documentos de operaciones que
puedan intervenir en la calidad del producto deben seguir procedimientos
escritos y constar como documentos en el archivo maestro:
a) "layout" de las instalaciones y
equipamientos;
b) procedimientos de limpieza y mantenimiento de las
instalaciones;
c) procedimientos de limpieza y mantenimiento de los
equipos;
d) instrucciones para el uso de los equipos;
e) procedimientos de calibración y aforado de los
equipos;
f) programas de entrenamiento del personal técnico;
g) procedimientos de devolución de materiales;
h) procedimientos para el rechazo de materiales;
i) procedimientos para el almacenamiento y
distribución;
j) procedimientos para reclamos;
k) otros.
A.3.11.3 Archivo histórico. El archivo histórico debe
existir para cada lote fabricado, en una forma ordenada, para mantener la
uniformidad en las informaciones, debiendo contener los siguientes documentos:
A.3.11.3.1. Con relación a la producción:
a) nombre del producto, número de lote;
b) número de lote de cada materia prima y material de
acondicionamiento utilizados en la confección del referido lote;
c) equipos utilizados y documentos relativos a la
preparación de los mismos, con fechas y firmas;
d) etapas detalladas de fabricación;
e) fecha, hora y firma de los responsables en las
etapas críticas;
f) registro de los parámetros del proceso de
esterilización y otros documentos relacionados con el lote;
g) resultado de todos los controles de proceso;
h) registro detallado de cualquier no conformidad y
firma responsable;
i) rótulo utilizado con el número de lote respectivo.
A.3.11.3.2. Con relación a Control de Calidad.
a) materias primas y material de acondicionamiento:
- registro completo conteniendo:
número de lote, fecha de los análisis, nombre de los materiales y sus
respectivos proveedores;
- informe de los resultados de los
análisis realizados, comparados con las especificaciones establecidas;
b) lote:
- registro completo describiendo los
ensayos de productos semielaborados y productos terminados con todos los datos
obtenidos en cada ensayo, incluyendo todos los gráficos, registros instrumentales,
cuando los hubiera:
- informe de los resultados de los
ensayos comparando con las especificaciones establecidas;
c) control ambiental:
- protocolo conforme al ítem A.3.2.4.
de esta recomendación.
d) control de agua:
- protocolo conforme al ítem A.3.4.3.
de esta recomendación.
A.3.11.3.3. Registros de recepción de materias
primas y de materiales de acondicionamiento:
a) nombre del material;
b) fecha de recepción;
c) nombre del proveedor;
d) lote del proveedor o número de referencia;
e) cantidad total o número de bultos recibidos;
f) número de lote adoptado por la empresa para la
identificación.
A.3.11.3.4. Registro de calibraciones y aforados
de los equipos. Conforme ítem A.3.3.6. de esta recomendación.
A.3.11.3.5. Registro de validación del proceso de
esterilización.
Conforme ítem A.3.7.2. de esta
recomendación.
A. 3.12. Reclamos.
A. 3.12.1. Procedimiento de admisión de los
reclamos. Deben ser establecidos y seguidos los procedimientos de admisión de
reclamos por escrito con relación a los productos. Tales procedimientos deben
incluir definiciones para la revisión por CC en cuanto a las especificaciones
de tales productos y a la necesidad de una investigación.
A.3.12.2. Registro de reclamos. Un registro
escrito de cada reclamo debe ser mantenido en un archivo propio en CC durante
el plazo de validez del producto, tal registro debe contener el nombre del
producto, número de lote, nombre del reclamante, naturaleza y respuesta del
reclamo.
Cuando fuera llevada a cabo una
investigación, los registros escritos deben incluir las conclusiones de la
investigación y los recaudos tomados. En el caso de no ser necesaria la
investigación, el registro escrito debe incluir la razón por la cual la
investigación fue considerada innecesaria y el nombre del responsable de tal
determinación.
A.3.13. Auditoría interna. Auditorías periódicas al
programa de garantía de calidad deben ser implementadas con el objeto de
verificar su observancia. Las auditorías deben ser realizadas de acuerdo con
procedimientos escritos, y por individuos entrenados, que no tengan relación
directa con el área a ser auditada. Los resultados y conclusiones de las
auditorías internas deben ser documentados por medio de informes escritos, que
deben ser revisados por la gerencia responsable de las áreas auditadas. Las
acciones correctivas o preventivas que se consideraran necesarias deben ser
acompañadas, si es necesario, por nuevas auditorías.
A.4. INSPECCIONES.
A.4.1. Inspecciones oficiales. El laboratorio
farmacéutico productor, estará sometido a inspecciones oficiales de acuerdo con
la guía para inspecciones (A-5) cuyas conclusiones deberán ser debidamente
documentadas y archivadas.
A.4.2. Inspecciones internas. Inspecciones para verificar
la observancia de los procedimientos de las buenas prácticas de fabricación de
las SPGV deberán ser realizadas periódicamente por la empresa, y sus
conclusiones debidamente documentadas y archivadas.
A.5. GUIA PARA INSPECCIONES
A.5.1. Administración e información general
|
|
Sí
|
No
|
Observaciones
|
|
A.5.1.1. ¿Cuál es la razón social de la empresa?
|
|
|
|
|
A.5.1.2. ¿Con quién fue hecho el contacto inicial?
|
|
|
|
|
A.5.1.3. ¿El farmacéutico responsable está presente?
|
|
|
|
|
A.5.1.4. ¿Existe prueba de su inscripción en el Organismo
Sanitario Nacional competente?
|
|
|
|
|
A.5.1.5. ¿Existe autorización del funcionamiento del
establecimiento por el Organismo Sanitario Nacional competente?
|
|
|
|
|
A.5.1.6. ¿La empresa posee autorización por Organismos
competentes para funcionamiento referente a la localización, protección
ambiental y seguridad de instalaciones?
|
|
|
|
|
A. 5.1.7. ¿Fueron exhibidos los planos de los edificios?
|
|
|
|
|
A.5.1.8. ¿Cuál es la superficie de terreno ocupado por la
empresa?
|
|
|
|
|
A.5.1.9. ¿Cuál es la superficie total ocupada por la
empresa?
|
|
|
|
|
A.5.1.10. ¿De cuántos edificios está compuesta la planta?
|
|
|
|
|
A.5.1.11. ¿Cuál es la superficie ocupada por cada
edificio?
|
|
|
|
|
A.5.1.12. ¿Cuál es el número de empleados que pertenecen a
la empresa?
|
|
|
|
|
A.5.1.13. ¿Cuál es el número de empleados que están
directamente ligados a operaciones de producción?
|
|
|
|
|
A.5.1.14. ¿ Fueron verificadas las libretas sanitarias de
los empleados?
|
|
|
|
|
A.5.1.15. ¿Fue exhibida la lista de los productos de
propiedad de la firma que están en fabricación y de los que no están?
|
|
|
|
|
A.5.1.16. ¿Todos esos productos están debidamente
registrados en el Organismo Sanitario Nacional competente?
|
|
|
|
|
A.5.1.17. ¿Cuál es la capacidad de producción del
establecimiento por forma farmacéutica y envase?
|
|
|
|
|
A.5.1.18. ¿Cuál es la capacidad de producción propia para
cada producto fabricado en la empresa?
|
|
|
|
|
A.5.1.19. ¿Cuál es la capacidad contratada a terceros para
cada producto?
OBSERVACION: Como parte de la inspección, deben
ser incluidas las empresas con las cuales se mantienen contratos de
producción, los productos involucrados y los volúmenes respectivos.
|
|
|
|
|
A.5.1.20. ¿Importa materia prima o producto terminado?
|
|
|
|
|
A.5.1.21. ¿Exporta materia prima o producto terminado?
|
|
|
|
|
A.5.2. Depósitos: Identificación. Debe llenarse una guía general y una
por cada depósito existente.
|
|
A.5.2.1. Condiciones externas.
|
|
A.5.2.1.1. ¿El aspecto externo del predio es bueno?
|
|
|
|
|
A.5.2.1.2. ¿Los alrededores del predio están
limpios?
|
|
|
|
|
A.5.2.1.3. ¿Existen protectores contra la
entrada de roedores, insectos y aves?
|
|
|
|
|
A.5.2.1.4.
¿Existen fuentes de
polución (industrias u otras) próximas al predio?
|
|
|
|
|
A.5.2.1.5. Si el depósito es un galpón, ¿las
condiciones de techado son buenas?
|
|
|
|
|
A.5.2.1.6. ¿Las vías de acceso al depósito son
buenas?
|
|
|
|
|
A.5.2.2. Condiciones Internas
|
|
A.5.2.2.1. ¿El predio es apropiado?
|
|
|
|
|
A.5.2.2.2.
¿Existen agujeros o
rajaduras en el predio?
|
|
|
|
|
A.5.2.2.3.
¿Es fácil de
limpiar?
|
|
|
|
|
A.5.2.2.4.
¿Las paredes están
bien conservadas (rajaduras, pintura descascarada)?
|
|
|
|
|
A.5.2.2.5.
¿El techo está en
buenas condiciones (goteras)?
|
|
|
|
|
A.5.2.2.6. ¿Existen cañerías sobre los
materiales almacenados, están en buen estado, no presentan filtraciones?
|
|
|
|
|
A.5.2.3. Aspectos generales
|
|
A.5.2.3.1. ¿La iluminación es suficiente?
|
|
|
|
|
A.5.2.3.2. ¿La ventilación es buena?
|
|
|
|
|
A.5.2.3.3. ¿Las instalaciones eléctricas están
en perfecto estado?
|
|
|
|
|
A.5.2.3.4. La temperatura se corresponde con
las condiciones de almacenamiento de los productos (medir y registrar la
temperatura).
|
|
|
|
|
A.5.2.3.5.
Si se necesita
humedad y temperatura controladas, ¿existen aparatos indicadores y registro
de esos datos?
|
|
|
|
|
A.5.2.3.6. ¿Existen indicios de la presencia
de roedores, insectos o aves en el local?
|
|
|
|
|
A.5.2.3.7. ¿Existen sistemas de combate de
roedores, insectos y aves?
|
|
|
|
|
A.5.2.3.8. ¿Es utilizado?
|
|
|
|
|
A.5.2.3.9. ¿Quién es el responsable? Nombre:
|
|
|
|
|
A.5.2.3.10. ¿Existen sanitarios en cantidad
suficiente?
|
|
|
|
|
A.5.2.3.11. ¿Están limpios?
|
|
|
|
|
A.5.2.3.12. ¿Existe un local separado para
refrigerio? ¿Está limpio?
|
|
|
|
|
A.5.2.3.13.
Existen vestuarios
en número suficiente?
|
|
|
|
|
A.5.2.3.14.
¿En el mismo
predio?
|
|
|
|
|
A.5.2.3.15. ¿Están limpios y en orden?
|
|
|
|
|
A.5.2.3.16.
¿El personal está
adecuadamente vestido y limpio?
|
|
|
|
|
A.5.2.3.17.
Si hubiera
necesidad de cámaras frigoríficas, ¿existen?
|
|
|
|
|
A.5.2.3.18. ¿La temperatura de la cámara está
controlada y registrada?
|
|
|
|
|
A.5.2.3.19. ¿Las balanzas son calibradas con
frecuencia?
|
|
|
|
|
A.5.2.3.20. ¿Existen registros de esas
calibraciones?
|
|
|
|
|
A.5.2.3.21. ¿La disposición del almacenamiento
es buena y racional, a fin de preservar la integridad e identidad de los
productos?
|
|
|
|
|
A.5.2.3.22. ¿Existen físicamente separadas para
materias primas, material de empaque, producto terminado y producto
semiterminado, si fuera el caso?
|
|
|
|
|
A.5.2.3.23. ¿Existe un área delimitada y
restringida para los materiales en cuarentena?
|
|
|
|
|
A.5.2.3.24. ¿Existe un área delimitada y
restringida para los materiales rechazados?
|
|
|
|
|
A.5.2.3.25. ¿Existe un área especial, aislada,
para el almacenamiento de rótulos?
|
|
|
|
|
A.5.2.3.26.
¿Existe un área
determinada y restringida para el almacenamiento de los medicamentos
devueltos?
|
|
|
|
|
A.5.2.3.27. ¿Existe un local para el
almacenamiento de productos inflamables y explosivos?
|
|
|
|
|
A.5.2.3.28. ¿Está situado en un área externa?
|
|
|
|
|
A.5.2.3.29. ¿Ofrece condiciones de seguridad?
|
|
|
|
|
A.5.2.3.30. ¿Existen depósitos, por separado y
seguros para sustancias corrosivas y cáusticas?
|
|
|
|
|
A.5.2.3.31.
¿Existen
recipientes para recolectar la basura?
|
|
|
|
|
A.5.2.3.32. ¿Están tapados?
|
|
|
|
|
A.5.2.3.33. ¿Son vaciados frecuentemente?
|
|
|
|
|
A.5.3.
Instalaciones de agua
|
|
A.5.3.1.
Agua de suministro.
|
|
A.5.3.1.1. ¿Cuál es el origen del agua usada
por la firma?
|
|
|
|
|
A.5.3.1.1.1. ¿De la red?
|
|
|
|
|
A.5.3.1.1.2. ¿De pozos surgentes o
semisurgentes?
|
|
|
|
|
A.5.3.1.1.3. ¿De otras fuentes de abastecimiento
(ríos, lagos, etc.)
|
|
|
|
|
A.5.3.1.2. ¿Sufre algún tratamiento antes de
ser almacenada?
|
|
|
|
|
A.5.3.1.3. ¿Cuál?
|
|
|
|
|
A.5.3.1.4. ¿Se hacen ensayos físicos, químicos
y bacteriológicos de esa agua?
|
|
|
|
|
A.5.3.1.5.
¿Existen registros?
|
|
|
|
|
A.5.3.1.6.
¿Las muestras son recogidas
en diversos puntos de la fábrica, inclusive los bebederos, para efectuar un
recuento bacteriano?
|
|
|
|
|
A.5.3.1.7. ¿Con qué frecuencia son hechos los
ensayos?
|
|
|
|
|
A.5.3.1.8. ¿A qué tratamientos se somete el
agua después de almacenada? Indicar secuencia.
|
|
|
|
|
A.5.3.1.9.
¿Cuántos tipos de
agua son utilizados en la planta? Enumerar:
|
|
|
|
|
A.5.3.2.
Tipo de agua. Debe
llenarse un formulario para cada tipo.
|
|
A.5.3.2.1. ¿La industria posee el equipamiento
necesario?
|
|
|
|
|
A.5.3.2.2.
¿Cuál es su
capacidad? .................litros/hora.
|
|
|
|
|
A.5.3.2.3. ¿Las resinas o membranas son
regeneradas o cambiadas cuando es necesario?
|
|
|
|
|
A.5.3.2.4. ¿Existen registros?
|
|
|
|
|
A.5.3.2.5.
¿Se hacen
diariamente ensayos físico-químicos?
|
|
|
|
|
A.5.3.2.6. ¿Existen registros?
|
|
|
|
|
A.5.3.2.7.
¿Con qué frecuencia
se hacen los ensayos bacteriológicos?
|
|
|
|
|
A.5.3.2.8.
¿Existen registros?
|
|
|
|
|
A.5.3.2.9. ¿Existe depósito para el agua
obtenida?
|
|
|
|
|
A.5.3.2.10. ¿Cuál es su capacidad?
|
|
|
|
|
A.5.3.3.6.
¿El agua producida
es utilizada inmediatamente? Sino ¿Por cuánto tiempo ella está almacenada? ¿A
qué temperatura? ¿Existe recirculación de esa agua?
|
|
|
A.5.3.3.7.
¿Existe algún
tratamiento para evitar la contaminación? ¿Cuál?
|
|
|
A.5.3.3.8. ¿Se hacen controles
físico-químicos? ¿Cuáles? ¿Con qué frecuencia? ¿Existen registros?
|
|
|
A.5.3.3.9. ¿Se hacen controles
bacteriológicos? ¿Con qué frecuencia? ¿Existen registros?
|
|
|
A.5.3.3.10. ¿Se hace control de piretógenos?
¿Con qué frecuencia? ¿Existen registros?
|
|
|
A.5.3.3.11. ¿El transporte o circulación de ese
agua se hace por cañería? Caso contrario ¿Cuál es el material y el sistema
utilizado? ¿De qué material es la cañería?
|
|
|
A.5.3.3.12. ¿Es liberada por Control de Calidad
antes de ser utilizada?
|
|
|
A.5.3.3.13. ¿Se hace sanitarización del
sistema? ¿Cómo? ¿Cuál es la frecuencia? ¿Existen registros?
|
|
|
A.5.3.3.14. ¿Existen procedimientos escritos de
sanitarización del sistema? ¿Son utilizados?
|
|
|
A.5.3.3.15.
¿Se hace
mantenimiento preventivo en los equipamientos del sistema? ¿Cuál es la
frecuencia? ¿Existen registros?
|
|
|
A.5.3.3.16. ¿El sistema de producción de agua
para inyectables está validado de forma de garantizar el cumplimiento de las
especificaciones establecidas por las ediciones vigentes de la Farmacopea Europea y de la Farmacopea de Estados Unidos de Norteamérica? ¿Existen registros?
|
|
|
A.5.4.
Recepción y almacenamiento
|
|
A.5.4.1.
Materias primas y
envases.
|
|
A.5.4.1.1. ¿Las materias primas son examinadas
cuando llegan al depósito para verificar si sufrieron daños durante el
transporte?
|
|
|
A.5.4.1.2.
¿Existen documentos
apropiados para su recepción?
|
|
|
A.5.4.1.3.
¿Son debidamente
completadas?
|
|
|
A.5.4.1.4. ¿Cada lote de materia prima recibe
un número de registro?
|
|
|
A.5.4.1.5. ¿Ese número acompaña a la materia
prima durante toda su utilización?
|
|
|
A.5.4.1.6. ¿Las materias primas son
conservadas en cuarentena hasta su liberación por el CC?
|
|
|
A.5.4.1.7. ¿Existen etiquetas apropiadas para
la identificación de la materia prima en cuarentena?
|
|
|
A.5.4.1.8.
¿Toda etiqueta
(identificación, cuarentena, aprobación, etc.) está fijada al cuerpo del
recipiente (no sobre la tapa)?
|
|
|
A.5.4.1.9.
¿Las materias primas son todas, sin excepción, muestreados por CC, de acuerdo
con sistemas apropiados y confiables?
|
|
|
A.5.4.1.10.
¿Si es aprobada, la materia prima recibe la etiqueta apropiada de
identificación y es inmediatamente llevada a las áreas normales de
almacenamiento?
|
|
|
A.5.4.1.11.
¿Las materias primas rechazadas son debidamente identificadas y aisladas?
|
|
|
A.5.4.1.12.
¿La disposición del almacenamiento es buena y racional, a fin de preservar la
integridad e identidad de las materias primas?
|
|
|
A.5.4.1.13.
¿Existen fichas de stock para cada material almacenado?
|
|
|
A.5.4.1.14.
¿Existe otro sistema adecuado de control del stock?
|
|
|
A.5.4.1.15.
¿Es funcional?
|
|
|
A.5.4.1.16.
¿Existen envases conteniendo material (latas, tambores, frascos, etc.) que
fueran dejados abiertos o semiabiertos)?
|
|
|
A.5.4.1.17.
¿El uso del material es hecho por orden de entrada, esto es, el más antiguo
en primer lugar?
|
|
|
A.5.4.1.18.
¿Los recipientes plásticos de las SPGV, son producidos en la fábrica?
|
|
|
A.5.4.1.19.
¿Los recipientes plásticos para las SPGV, se proveen cerrados por
termosellado?
|
|
|
A.5.4.1.20.
¿Existen recipientes para residuos? ¿Están identificados? ¿Están bien
cerrados? ¿Se vacían con frecuencia?
|
|
|
A.5.4.2.
Material de empaque.
|
|
|
A.5.4.2.1.
¿El material de empaque es examinado visualmente cuando llega para verificar
si sufrió daños durante su transporte?
|
|
|
A.5.4.2.2.
¿Existen documentos apropiados para su recepción?
|
|
|
A.5.4.2.3.
Están debidamente completados?
|
|
|
A.5.4.2.4.
¿El material de empaque es conservado en cuarentena hasta su liberación por
CC?
|
|
|
A.5.4.2.5.
¿Existen etiquetas apropiadas de identificación del material de empaque en
cuarentena?
|
|
|
A.5.4.2.6.
¿Los materiales de empaque son todos, sin excepción, muestreados por CC de
acuerdo con sistemas apropiados y confiables?
|
|
|
A.5.4.2.7.
¿Si el material de empaque ha sido aprobado, recibe etiqueta de
identificación y es llevado inmediatamente para las áreas normales de
almacenamiento?
|
|
|
A.5.4.2.8.
¿Los materiales de empaque rechazados son claramente identificados y aislados
en locales apropiados?
|
|
|
A.5.4.2.9.
¿La disposición del almacenamiento es buena y racional, a fin de preservar la
integridad e identidad de los materiales?
|
|
|
A.5.4.2.10.
¿Existen fichas de stock para cada material almacenado?
|
|
|
A.5.4.2.11.
¿Existe otro sistema adecuado de control de stock?
|
|
|
A.5.4.2.12.
¿Es funcional?
|
|
|
A.5.4.2.13.
¿Existe un área destinada al almacenamiento de rótulos?
|
|
|
A.5.4.2.14.
¿Es permitida la entrada al área solamente a las personas autorizadas?
|
|
|
A.5.4.2.15.
¿El uso del material se hace por orden de llegada, o sea, el más antiguo en
primer lugar?
|
|
|
A.5.4.2.16.
¿Existen embalajes que fueron dejados abiertos, semi abiertos o mal
acondicionados?
|
|
|
A.5.4.2.17.
¿Existen recipientes para residuos? ¿Están identificados? ¿Están bien
cerrados? ¿Se vacían con frecuencia?
|
|
|
A.5.4.3.
Producto terminado.
|
|
|
A.5.4.3.1.
¿Están almacenados en área física propia, separados de otros materiales?
|
|
|
A.5.4.3.2.
¿Existen áreas de cuarentena para productos terminados que están esperando la
aprobación de CC?
|
|
|
A.5.4.3.3.
¿Los productos están identificados como tal (en análisis o cuarentena)?
|
|
|
A.5.4.3.4.
¿Existe un sistema eficaz que evite la expedición de productos en cuarentena?
|
|
|
A.5.4.3.5.
¿El local está limpio?
|
|
|
A.5.4.3.6.
¿Está bien ventilado?
|
|
|
A.5.4.3.7.
¿Está bien iluminado?
|
|
|
A.5.4.3.8.
¿Existen indicadores de temperatura y humedad?
|
|
|
A.5.4.3.9.
¿Funcionan?
|
|
|
A.5.4.3.10.
¿Existen registros de estas condiciones ambientales?
|
|
|
A.5.4.3.11.
¿Se mantienen registros de productos recibidos?
|
|
|
A.5.4.3.12.
¿Se mantienen registros de productos expedidos (o despachados)?
|
|
|
A.5.4.3.13.
¿La salida de los productos del área obedece a un orden cronológico de
entrada?
|
|
|
A.5.4.3.14.
¿Los productos están lo suficientemente separados entre sí, a fin de evitar
posibles mezclas entre lotes o productos?
|
|
|
A.5.4.3.15.
¿Los productos son apilados con seguridad?
|
|
|
A.5.4.3.16.
¿Los productos están almacenados en contacto directo con el suelo?
|
|
|
A.5.4.3.17.
¿Hay espacio adecuado entre los productos y las paredes para facilitar la
inspección?
|
|
|
A.5.4.3.18.
¿Las áreas están protegidas contra la entrada de roedores, insectos y aves?
|
|
|
A.5.4.3.19.
¿Hay un programa de control de roedores, insectos y aves?
|
|
|
A.5.4.3.20.
¿Existe un local para el almacenamiento de productos que necesiten bajas
temperaturas?
|
|
|
A.5.4.3.21.
¿Las superficies del piso, paredes y techo son de fácil limpieza?
|
|
|
A.5.4.3.22.
¿Todos los productos almacenados tienen claramente identificado el número de
lote en sus cajas y envases?
|
|
|
A.5.4.3.23.
¿Todos los productos almacenados tienen claramente identificada la fecha de
vencimiento en sus cajas y envases?
|
|
|
A.5.4.3.24.
¿Todos los productos almacenados están dentro de su plazo de validez?
|
|
|
A.5.4.3.25.
¿Los productos vencidos son inmediatamente retirados del stock?
|
|
|
A.5.4.3.26.
¿Existe un control de distribución a nivel primario de los productos
terminados, de modo de permitir la identificación de su destino, y de ser
necesario, su recuperación?
|
|
|
A.5.4.4.
Devoluciones y/o retiro del mercado.
|
|
|
A.5.4.4.1.
¿Existe un protocolo de devoluciones que acompaña al producto devuelto?
|
|
|
A.5.4.4.2.
¿Existe un área específicamente determinada para la recepción y
almacenamiento de los productos devueltos o retirados?
|
|
|
A.5.4.4.3.
¿Esos productos son identificados como tales?
|
|
|
A.5.4.4.4.
¿Se toman recaudos especiales por parte de las personas responsables con
relación a esas devoluciones?
|
|
|
A.5.4.4.5.
Se avisa inmediatamente a CC luego de recibir un producto devuelto o
retirado?
|
|
|
A.5.4.4.6.
¿Se toman recaudos inmediatos para destruir o recuperar el lote en cuestión,
conforme sea la decisión de CC?
|
|
|
A.5.4.4.7.
¿Las personas responsables son avisadas de los problemas ocurridos, para que
sean tomados los debidos recaudos, y no se repita en otros lotes del mismo
producto?
|
|
|
A.5.4.4.8.
¿Los resultados de las inspecciones y análisis son registrados?
|
|
|
A.5.4.4.9.
¿Todas las decisiones tomadas son debidamente registradas?
|
|
|
A.5.4.4.10.
¿La empresa establece y mantiene procedimientos de retiro del producto del
mercado?
|
|
|
A.5.4.4.11.
¿Existe una persona responsable designada para la coordinación y ejecución
del procedimiento de retiro?
|
|
|
A.5.4.4.12.
¿Se mantienen informes de los retiros de productos del mercado, así como de
sus causas?
|
|
|
A.5.4.4.13.
En caso de retiro de productos por desvíos de calidad, ¿las autoridades
competentes de los demás países son informadas inmediatamente?
|
|
|
A.5.4.4.14.
¿Los registros de distribución a nivel primario, de los productos, quedan
disponibles para una pronta acción de retiro del mercado?
|
|
|
A.5.4.4.14.1.
¿Esos registros contienen información que permitan el rastreo y determinación
de cuáles son los destinatarios resultantes de la distribución primaria?
|
|
|
A.5.4.4.15.
¿Existen informes concluyentes sobre todo proceso para cada producto retirado
del mercado y su destino?
|
|
|
A.5.5.
Producción
|
|
|
A.5.5.1.
Personal.
|
|
|
A.5.5.1.1.
¿Quién es responsable de la producción?
|
|
|
A.5.5.1.2.
¿Cuál es su nivel profesional?
|
|
|
A.5.5.1.3.
¿El responsable posee calificación y competencia necesaria para esa función?
|
|
|
A.5.5.1.4.
¿Existe un organigrama?
|
|
|
A.5.5.1.5.
¿El personal técnico especializado es suficiente?
|
|
|
A.5.5.1.6.
¿Existe un programa de instrucción del personal?
|
|
|
A.5.5.1.7.
¿Toda admisión de personal es precedida de examen médico?
|
|
|
A.5.5.1.8.
¿Ese examen es repetido periódicamente?
|
|
|
A.5.5.1.9.
¿El personal, cuyo estado de salud sea dudoso, es apartado inmediatamente de
su lugar de trabajo, hasta que esté recuperado?
|
|
|
A.5.5.2.
Condiciones generales de las áreas de producción (un formulario por predio)
|
|
|
A.5.5.2.1.
¿El predio se encuentra externamente en buen estado?
|
|
|
A.5.5.2.2.
¿Los alrededores del predio están limpios y sin basura?
|
|
|
A.5.5.2.3.
¿El techo, las paredes y las ventanas están en orden?
|
|
|
A.5.5.2.4.
¿Existen industrias que produzcan polución en los alrededores del predio?
|
|
|
A.5.5.2.5.
¿Existe protección contra la entrada de roedores, insectos y aves?
|
|
|
A.5.5.2.6.
¿Las áreas de producción están limpias?
|
|
|
A.5.5.2.7.
¿Existe un programa de limpieza por escrito?
|
|
|
A.5.5.2.8.
¿Está prohibido comer, beber y fumar en las áreas de producción?
|
|
|
A.5.5.2.9.
¿Existen vestuarios en cantidad suficiente?
|
|
|
A.5.5.2.10.
¿Existen sanitarios en cantidad suficiente?
|
|
|
A.5.5.2.11.
¿Los sanitarios y vestuarios están limpios y bien arreglados?
|
|
|
A.5.5.2.12.
¿Existe cantina y/o comedor?
|
|
|
A.5.5.2.13.
¿Existen normas de seguridad por escrito? ¿Son cumplidas?
|
|
|
A.5.5.2.14.
¿Los extintores y el sistema de agua para incendios están correctamente
localizados?
|
|
|
A.5.5.2.15.
¿El personal recibe ropas y zapatos apropiados para el trabajo?
|
|
|
A.5.5.2.16.
¿En las áreas de producción entran solamente personas con ropas apropiadas?
|
|
|
A.5.5.2.17.
¿Los pisos son adecuados para cada área particular de trabajo?
|
|
|
A.5.5.2.18.
¿Las ventanas poseen alambre tejido contra la entrada de insectos y aves?
|
|
|
A.5.5.2.19.
¿Existe un programa de combate contra roedores, insectos y aves?
|
|
|
A.5.5.2.20.
¿La circulación interna es adecuada?
|
|
|
A.5.5.2.21.
¿La iluminación de las áreas de circulación es suficiente?
|
|
|
A.5.5.2.22.
¿La ventilación de las áreas de circulación es buena?
|
|
|
A.5.5.2.23.
¿El sistema de sumidero es adecuado?
|
|
|
A.5.5.2.24.
¿Las rejillas poseen sifones?
|
|
|
A.5.5.2.25.
¿Son desinfectados frecuentemente?
|
|
|
A.5.5.2.26.
¿Las instalaciones eléctricas están en buenas condiciones?
|
|
|
A.5.5.2.27.
¿Las cañerías de agua, vapor, gas, aire comprimido, electricidad, etc., están
debidamente identificadas?
|
|
|
A.5.5.2.28.
¿Existen líneas muertas?
|
|
|
A.5.5.2.29.
¿Las paredes y techos están revestidos de material fácilmente lavable?
|
|
|
A.5.5.2.30.
¿Las paredes, techos y pisos no tiene rajaduras, ni presentan la pintura
descascarada?
|
|
|
A.5.5.2.31.
¿Los recipientes para la recolección de basura, existentes en diversos
sectores, están bien tapados?
|
|
|
A.5.5.2.32.
¿Son vaciados frecuentemente?
|
|
|
A.5.5.2.33.
¿Cuál es el área, en metros cuadrados, ocupados para la producción
farmacéutica, excluido el depósito?
|
|
|
A.5.5.2.34.
¿Cuál es el número de operarios del sector de producción?
|
|
|
A.5.5.2.35.
¿Cuál es la relación área- operario?
|
|
|
A.5.5.2.36.
¿Existe un organigrama?
|
|
|
A.5.5.2.37.
¿Existe un sistema para controlar la entrada de personas no autorizadas en el
área de producción?
|
|
|
A.5.5.3.
Sector de pesada:
|
|
|
A.5.5.3.1.
¿El área está físicamente separada de las demás dependencias por paredes, u
otro tipo de separación?
|
|
|
A.5.5.3.2.
¿Los materiales usados para las pesadas (recipientes, cucharones, espátulas,
pipetas, etc.) están limpias?
|
|
|
A.5.5.3.3.
¿Estos materiales son guardados en lugar limpio?
|
|
|
A.5.5.3.4.
¿Las balanzas son calibradas regularmente?
|
|
|
A.5.5.3.5.
¿Existen registros de esas calibraciones?
|
|
|
A.5.5.3.6.
¿Son usados equipos de protección (guantes, gorros, máscaras, etc.) cuando
son necesarios durante las pesadas?
|
|
|
A.5.5.3.7.
¿Los recipientes que contienen la materia prima a ser pesada, son limpiados
antes de ser abiertos?
|
|
|
A.5.5.3.8.
¿Después de la pesada, esos recipientes son bien cerrados?
|
|
|
A.5.5.3.9.
¿Los materiales, después de pesados, son identificados, a fin de evitar
mezclas?
|
|
|
A.5.5.3.10.
En esta identificación constan:
|
|
|
A.5.5.3.10.1.
Nombre y número de lote del producto al que se destina la materia prima
|
|
|
A.5.5.3.10.2.
Nombre y número de lote de la materia prima
|
|
|
A.5.5.3.10.3.
Cantidad que fue pesada.
|
|
|
A.5.5.3.10.4.
Control de pesada con el visto bueno del operario que pesó y del que verificó
la pesada.
|
|
|
A.5.5.3.11.
¿Los operadores están con uniformes limpios?
|
|
|
A.5.5.3.12.
¿El área es ventilada adecuadamente?
|
|
|
A.5.5.3.13.
¿Posee algún sistema de extracción?
|
|
|
A.5.5.3.14.
El área está iluminada adecuadamente?
|
|
|
A.5.5.3.15.
¿Las materias primas de un lote, ya pesadas, son separadas físicamente de las
de los otros lotes?
|
|
|
A.5.5.3.16.
¿El área posee un local para el lavado del material utilizado?
|
|
|
A.5.5.3.17.
¿Los recipientes usados en la pesada de materia prima, si son usados en
nuevas pesadas, son lavados perfectamente y libres de cualquier
identificación anterior?
|
|
|
A.5.5.3.18.
¿Las materias primas más antiguas son usadas en primer lugar?
|
|
|
A.5.5.4.
Sector de preparación de soluciones:
|
|
|
A.5.5.4.1.
¿El área ocupada es adecuada para el volumen de las operaciones?
|
|
|
A.5.5.4.2.
¿Cuál es el área, en metros cuadrados, ocupada por el sector?
|
|
|
A.5.5.4.3.
¿Cuál es el número de operarios existente en el sector?
|
|
|
A.5.5.4.4.
¿Cuál es la relación área-operario?
|
|
|
A.5.5.4.5.
¿La distribución de los equipos es ordenada y racional?
|
|
|
A.5.5.4.6.
¿Todos usan gorros?
|
|
|
A.5.5.4.7.
¿Cuando es necesario, usan guantes, máscaras y anteojos de protección?
|
|
|
A.5.5.4.8.
¿Existe en el local un sistema de renovación de aire?
|
|
|
A.5.5.4.9.
¿Existe un procedimiento de fabricación (ficha técnica de fabricación FTF) a
ser seguido, que represente una copia fiel de la fórmula patrón (de la ficha
de producción)?
|
|
|
A.5.5.4.10.
¿Las instrucciones contenidas en la fórmula de fabricación son seguidas con
exactitud?
|
|
|
A.5.5.4.11.
¿Cada fase crítica de la producción lleva la firma del operador y del
supervisor inmediato?
|
|
|
A.5.5.4.12.
¿Todos los recipientes usados en la producción de un lote están identificados
de acuerdo con su contenido y número de solución o sublote, a fin de evitar
mezclas?
|
|
|
A.5.5.4.13.
¿Todo equipo usado en la fabricación de un lote está etiquetado exactamente
como los recipientes?
|
|
|
A.5.5.4.14.
¿Después de su uso, todos los utensilios, equipos y recipientes son bien
lavados y si es necesario, esterilizados y conservados así, hasta ser
nuevamente usados?
|
|
|
A.5.5.4.15.
¿Son identificados por etiquetas que aseguran esa condición?
|
|
|
A.5.5.4.16.
¿Existe una adecuada separación física entre los equipos para evitar mezcla o
contaminación cruzada, cuando hay fabricación simultánea de lotes de
productos diferentes?
|
|
|
A.5.5.4.17.
¿El agua usada en la fabricación es previamente analizada y liberada por CC?
|
|
|
A.5.5.4.18.
¿Son efectuados controles durante el proceso de fabricación a fin de
garantizar la calidad del lote?
|
|
|
A.5.5.4.19.
¿Existen registros, por escrito, de esos controles?
|
|
|
A.5.5.4.20.
¿Los tanques conteniendo el producto a ser envasado están bien tapados e
identificados apropiadamente con, como mínimo los siguientes datos:
|
|
|
A.5.5.4.20.1.
Nombre del producto.
|
|
|
A.5.5.4.20.2.
Número de lote o sublote.
|
|
|
A.5.5.4.20.3.
Volumen de la solución contenida en el tanque.
|
|
|
A.5.5.4.20.4.
¿Los tanques conteniendo la solución a ser envasada son manipulados de modo
de garantizar la no contaminación del producto?
|
|
|
A.5.5.5.
Sector de lavado de recipientes de vidrio
|
|
|
A.5.5.5.1.
¿Existe un local separado para el lavado y esterilización de recipientes de
vidrio vacío?
|
|
|
A.5.5.5.2.
¿El área ocupada es adecuada para el volumen de las operaciones?
|
|
|
A.5.5.5.3.
¿La existencia y distribución de los equipamientos es ordenada, racional y
adecuada al volumen de operaciones?
|
|
|
A.5.5.5.4.
¿El área de circulación está libre de obstáculos?
|
|
|
A.5.5.5.5.
¿Los uniformes utilizados son adecuados? ¿Están limpios y en buenas
condiciones?
¿Se usan fuera de planta?
|
|
|
A.5.5.5.6.
¿Las máquinas de lavado de recipientes de vidrio poseen presión suficiente
para cumplir su finalidad?
|
|
|
A.5.5.5.7.
¿Cuál es el tipo de agua utilizada en la alimentación de las máquinas de
lavado de frascos?
|
|
|
A.5.5.5.8.
¿Existe algún tipo de filtro en el sistema de lavado de frascos?
|
|
|
A.5.5.5.9.
¿Las estufas de secado funcionan perfectamente y poseen indicadores de
temperatura?
|
|
|
A.5.5.5.10.
¿Los recipientes lavados y secos son transferidos con seguridad al área de
envase evitando una posible contaminación?
|
|
|
A.5.5.5.11.
¿Los recipientes lavados y secos son debidamente identificados?
|
|
|
A.5.5.6.
Sectores de llenado, esterilización e inspección
|
|
|
A.5.5.6.1.
¿Existen áreas de ambiente controlado para el llenado de los envases?
|
|
|
A.5.5.6.1.1.
Indicar clase de área de la sala de llenado.
|
|
|
A.5.5.6.1.2.
Indicar clase de área de la línea de llenado.
|
|
|
A.5.5.6.2.
¿El área ocupada concuerda con el volumen de las operaciones?
|
|
|
A.5.5.6.3.
¿La distribución de los equipos es ordenada y racional?
|
|
|
A.5.5.6.4.
¿El personal está adecuadamente uniformado y limpio?
|
|
|
A.5.5.6.5.
¿Todos usan gorros, guantes y máscaras?
|
|
|
A.5.5.6.6.
Consignar condiciones de sala de llenado:
a) Ventilación
b) Temperatura
c) Humedad
|
|
|
A.5.5.6.7.
¿Existe sistema de aire comprimido?
|
|
|
A.5.5.6.8.
¿Existen filtros para la entrada de aire?
|
|
|
A.5.5.6.9.
¿La eficiencia de los filtros es verificada con frecuencia?
|
|
|
A.5.5.6.10.
¿La sala posee presión positiva de aire?
|
|
|
A.5.5.6.11.
¿Son hechos controles frecuentes del volumen de llenado de los recipientes?
|
|
|
A.5.5.6.12.
¿El CC verifica frecuentemente el volumen de los recipientes?
|
|
|
A.5.5.6.13.
¿Existe registro, por escrito, de esas verificaciones?
|
|
|
A.5.5.6.14.
¿La entrada del personal en el área es debidamente controlada?
|
|
|
A.5.5.6.15.
¿Si las unidades carecen de identificación, los carros que contienen
recipientes ya llenos están debidamente identificados?
|
|
|
A.5.5.6.16.
¿Los autoclaves están identificados adecuadamente?
|
|
|
A.5.5.6.17.
¿Existen registradores de presión y temperatura?
|
|
|
A.5.5.6.18.
¿Existen registros, por escrito, de esos datos?
|
|
|
A.5.5.6.19.
¿Existen instrucciones por escrito, sobre el tiempo y temperatura de
autoclavado?
|
|
|
A.5.5.6.20.
¿Se realizan, periódicamente, ensayos físicos o biológicos para la
verificación del funcionamiento de los autoclaves?
|
|
|
A.5.5.6.21.
¿Existen registros por escrito?
|
|
|
A.5.5.6.22.
¿Después del autoclavado se hace algún ensayo para verificar si los envases
están bien cerrados?
|
|
|
A.5.5.6.23.
¿El área ocupada por los autoclaves concuerda con el volumen de las
operaciones?
|
|
|
A.5.5.6.24.
¿La distribución de los equipos es ordenada y racional?
|
|
|
A.5.5.6.25.
¿El personal está uniformado adecuadamente?
|
|
|
A.5.5.6.26.
¿Todos usan gorros, y cuando es necesario, guantes especiales?
|
|
|
A.5.5.6.27.
¿Existe algún método que identifique los productos a ser esterilizados, y los
ya esterilizados?
|
|
|
A.5.5.6.28.
¿Existe sala especial para inspección de recipientes llenos?
|
|
|
A.5.5.6.29.
¿Los carros que contienen los envases ya inspeccionados son debidamente
identificados?
|
|
|
A.5.5.7.
Empaque
|
|
|
A.5.5.7.1.
¿Existe un local especial para el empaque final de los productos?
|
|
|
A.5.5.7.2.
¿El área ocupada concuerda con el volumen de las operaciones?
|
|
|
A.5.5.7.3.
¿La separación entre las líneas de empaque es suficiente, para evitar mezclas
de productos diferentes o lotes diferentes de un mismo producto?
|
|
|
A.5.5.7.4.
¿El área de circulación está libre?
|
|
|
A.5.5.7.5.
¿El personal está uniformado adecuadamente?
|
|
|
A.5.5.7.6.
¿Todos usan gorros, y cuando es necesario, guantes, máscaras y anteojos de
protección?
|
|
|
A.5.5.7.7.
¿La ventilación del local es suficiente?
|
|
|
A.5.5.7.8.
¿Las condiciones de seguridad del local son buenas?
|
|
A.5.5.7.9.
¿Las líneas de empaque
son verificadas, en cuanto a la presencia de material sobrante de productos
anteriores, antes de iniciar las operaciones?
A.5.5.7.10. ¿Los carros que contienen los
productos a ser embalados, están debidamente identificados en cuanto a su
contenido?
A.5.5.7.11.
¿Esos carros se
mantienen tapados y protegidos de la luz (si es necesario), durante todo el
proceso, y son abiertos cuando es necesario?
A.5.5.7.12. ¿Los carros conteniendo productos
diferentes se mantienen separados?
A.5.5.7.13. ¿Todo material de empaque a ser usado
tiene registro de aprobación del CC?
|
|
|
|
|
|
A.5.5.7.14. ¿Cada línea de empaque, está
visiblemente identificada, de acuerdo con el producto que está siendo
empacado?
|
|
|
|
|
A.5.5.7.15. ¿Se verifica la relación entre el
rendimiento teórico y el rendimiento real?
|
|
|
|
|
A.5.5.7.16. ¿Cualquier discrepancia es
registrada por escrito?
|
|
|
|
|
A.5.5.7.17. ¿Los productos empacados aguardan
en cuarentena hasta liberación por CC?
|
|
|
|
|
A.5.5.8. Rotulado
|
|
A.5.5.8.1.
¿El acceso a los
rótulos es permitido únicamente a personal debidamente autorizado?
|
|
|
|
|
A.5.5.8.2.
¿Los rótulos son
inspeccionados individualmente antes de ser entregados a la línea de empaque?
|
|
|
|
|
A.5.5.8.3.
¿Las máquinas de
rotular son examinadas antes de su uso, para verificar que no existan rótulos
del producto anterior?
|
|
|
|
|
A.5.5.8.4.
¿Los rótulos que
están en exceso o sellados se destruyen luego de terminado el empaque?
|
|
|
|
|
A.5.5.8.5. ¿El responsable de guardar y
almacenar los rótulos, verifica la cantidad devuelta y los almacena
cuidadosamente, a fin de evitar mezclas?
|
|
|
|
|
A.5.5.9.
Registros de
producción
|
|
A.5.5.9.1.
Formulario patrón
|
|
|
|
|
A.5.5.9.1.1.
¿Existe un
formulario patrón para cada producto a ser fabricado?
|
|
|
|
|
A.5.5.9.1.2.
¿Ese formulario
patrón está preparado y firmado por una persona responsable y endosado por
otra persona también responsable y competente?
|
|
|
|
|
A.5.5.9.1.3.
El formulario
patrón contiene:
|
|
|
|
|
A.5.5.9.1.3.1. ¿El nombre, la fórmula y la
presentación farmacéutica del producto?
|
|
|
|
|
A.5.5.9.1.3.2. ¿La cantidad teórica del producto a
ser producido?
|
|
|
|
|
A.5.5.9.1.3.3.
¿El nombre y
cantidad de cada materia prima como sus códigos?
|
|
|
|
|
A.5.5.9.1.3.4.
¿Instrucciones
detalladas de las etapas de fabricación, con indicación del equipamiento a
ser usado?
|
|
|
|
|
A.5.5.9.1.3.5. ¿Instrucciones adecuadas para el
rotulado y empaque del producto?
|
|
|
|
|
A.5.5.9.1.4. ¿Si fuera necesario modificar el
formulario patrón original, se siguen procedimientos racionales escritos para
tal fin?
|
|
|
|
|
A.5.5.9.2.
Ordenes de
producción
|
|
|
|
|
A.5.5.9.2.1.
¿Existen órdenes de
producción para cada lote fabricado?
|
|
|
|
|
A.5.5.9.2.2.
Estas órdenes
contienen:
|
|
|
|
|
A.5.5.9.2.2.1. ¿Instrucciones detalladas de las
etapas de fabricación con lugares apropiados para la firma de los
responsables e indicaciones de equipamiento a ser usado?
|
|
|
|
|
A.5.5.9.2.3.
¿Es registrado el
número de lote del producto?
|
|
|
|
|
A.5.5.9.2.4. ¿Es registrado el número del lote
de las materias primas a ser usadas?
|
|
|
|
|
A.5.5.9.2.5. ¿Es calculada la cantidad de
materia prima de acuerdo con su pureza?
|
|
|
|
|
A.5.5.9.2.6.
¿Existe un
responsable de ese cálculo?
|
|
|
|
|
A.5.5.9.2.7.
¿Ese cálculo es
ratificado por otro responsable?
|
|
|
|
|
A.5.5.9.2.8.
¿Se consigna
cálculo de rendimiento real obtenido en las diversas etapas de fabricación,
con relación al rendimiento teórico?
|
|
|
|
|
A.5.5.9.2.9.
¿Si fuera necesario
modificar las instrucciones éstas son registradas en lugar apropiado y
aprobadas por la persona competente?
|
|
|
|
|
A.5.5.9.2.10. ¿Luego de terminado el proceso de
producción, toda la documentación sobre el lote producido (hoja de
producción, rótulo, resultados analíticos, etiquetas, gráficos de autoclavado
de componentes y producto final, etc.) es archivada para referencia futura?
|
|
|
|
|
A.5.6.
Control de Calidad
|
|
A.5.6.1.
¿Existe en la
empresa un laboratorio de CC?
|
|
|
|
|
A.5.6.2. ¿Existe un organigrama del
laboratorio de CC?
|
|
|
|
|
A.5.6.3.
¿El CC es
responsable de aprobar o rechazar materias primas, productos intermedios y
sus contenedores, productos terminados, material de envase y empaque?
|
|
|
|
|
A.5.6.4. ¿A quién se reporta el responsable
del CC?
|
|
|
|
|
A.5.6.5.
¿Cuál es el nivel
profesional del responsable del CC?
|
|
|
|
|
A.5.6.6.
¿El CC está
equipado para ejercer su función?
|
|
|
|
|
A.5.6.7. ¿Posee personal técnico calificado
para ejercer su función?
|
|
|
|
|
A.5.6.8.
¿El área es adecuada?
|
|
|
|
|
A.5.6.9. ¿Posee un lugar adecuado para
equipos sensibles?
|
|
|
|
|
A.5.6.10.
¿Se hacen ensayos
biológicos?
|
|
|
|
|
A.5.6.11.
¿Posee una sala
especial para ensayos de esterilidad y dosajes microbiológicos?
|
|
|
|
|
A.5.6.12. ¿Posee bioterio?
|
|
|
|
|
A.5.6.12.1. ¿Está localizado dentro o fuera del
predio?
|
|
|
|
|
A.5.6.12.2. ¿Dentro del predio, las
instalaciones de aire acondicionado están totalmente separadas de cualquier
otro sistema?
|
|
|
|
|
A.5.6.12.3. ¿Existen registros de las
condiciones ambientales del bioterio?
|
|
|
|
|
A.5.6.12.4. ¿La limpieza del local es buena?
|
|
|
|
|
A.5.6.12.5. ¿Los animales están bien alojados,
bien alimentados y gozan de buena salud?
|
|
|
|
|
A.5.6.13. ¿Existe un procedimiento escrito
para el mantenimiento preventivo y la calibración de los aparatos utilizados?
|
|
|
|
|
A.5.6.14. ¿El programa es cumplido?
¿Existen registros?
|
|
|
|
|
A.5.6.15. ¿El CC mantiene registros por
escrito, de todos los ensayos efectuados?
|
|
|
|
|
A.5.6.16. ¿Son reservadas contramuestras de
las materias primas empleadas?
|
|
|
|
|
¿Está
definido el tiempo de conservación?
|
|
|
|
|
A.5.6.17. ¿Se conservan muestras de
referencia de cada lote de producto terminado, en su envase final?
¿Por cuánto tiempo?
|
|
|
|
|
A.5.6.18. ¿Existen funcionarios de CC
designados especialmente para el control de producción (inspectores de
producción)?
|
|
|
|
|
A.5.6.19. ¿Todos los ensayos necesarios
(materias primas, material de empaque, semi-terminados y finales), son
realizados por CC?
|
|
|
|
|
A.5.6.20. ¿Se verifica que cada lote de
producto esté de acuerdo con las especificaciones establecidas, antes de ser
liberado?
|
|
|
|
|
A.5.6.20.1.
¿CC verifica toda
la documentación del proceso?
|
|
|
|
|
A.5.6.21. ¿CC tiene especificaciones escritas
para todas las materias primas, material de empaque, productos intermedios y
productos finales?
|
|
|
|
|
|
|
|
|
|
A.5.6.22. ¿CC tiene por escrito todos los
procedimientos de muestreo y métodos analíticos utilizados?
A.5.6.23. ¿Existen instalaciones de seguridad
en el laboratorio (duchas, lavaojos, etc.)?
A.5.6.23.1. ¿Existe un programa de verificación
de esos equipamientos?
A.5.6.24. ¿Existe algún procedimiento con
relación a patrones de referencia y su mantenimiento?
A.5.6.25. ¿Los procedimientos usados por CC son
suficientes para asegurar la calidad de los lotes fabricados?
A.5.6.26. ¿Existen controles efectuados por
laboratorios contratados?
¿Qué
controles?
¿Existen
contratos?
A.5.6.27. ¿CC es responsable de aprobar o
rechazar productos elaborados, acondicionados o mantenidos bajo contrato por
terceros?
|
|
|
|
|
|
A.5.7.
Garantía de Calidad
|
|
A.5.7.1.
¿Existe en la
empresa un programa de garantía de calidad?
|
|
|
|
|
A.5.7.1.1. ¿Se divulga a todos los niveles?
|
|
|
|
|
A.5.7.2. ¿Existen normas escritas para la
divulgación y cumplimiento de buenas prácticas de fabricación?
|
|
|
|
|
A.5.7.2.1. ¿Se siguen estas normas?
|
|
|
|
|
A.5.7.3.
¿Existe en la
empresa un área que coordine las actividades de Garantía de Calidad?
|
|
|
|
|
A.5.7.4.
¿Están claramente
definidas las responsabilidades por la gestión de calidad?
|
|
|
|
|
A.5.7.5. ¿Existen procedimientos escritos o
sistemas para avalar la efectividad y aplicabilidad de las normas y sistemas
de Garantía de Calidad?
|
|
|
|
|
A.
5.7.6. ¿Existe un
programa de entrenamiento del personal?
|
|
|
|
|
A.5.7.6.1. ¿Se llevan registros del
entrenamiento de cada funcionario?
|
|
|
|
|
A.5.7.7.
¿Se desarrollan y
proyectan los productos de acuerdo con requisitos de buenas prácticas de
fabricación?
|
|
|
|
|
A.5.7.8.
¿Las operaciones de
producción y control están claramente definidas y escritas?
|
|
|
|
|
A.5.7.9.
¿Se entrenan los
funcionarios de modo de garantizar una correcta y completa ejecución de los
procesos y procedimientos definidos?
|
|
|
|
|
A.5.7.10. ¿Los nuevos conocimientos
adquiridos en los procesos o las adaptaciones y mejoras se implementan solo
después de una completa evaluación y aprobación?
|
|
|
|
|
A.5.7.11. ¿Se realizan autoinspecciones
periódicas para verificar el cumplimiento de buenas prácticas de fabricación?
|
|
|
|
|
A.5.7.11.1. ¿Existen registros?
|
|
|
|
|
A.5.7.12.
¿Existe un programa
escrito de estudio de la estabilidad de los productos con condiciones de los
ensayos, registros de los resultados, métodos analíticos usados, condiciones
de conservación de las muestras, envases primarios, periodicidad de análisis
y fecha de vencimiento?
|
|
|
|
|
A.5.7.12.1.
¿Se sigue el
programa?
|
|
|
|
|
A.5.7.13. ¿Existe un sistema o procedimiento
que permita verificar si se cumplen las condiciones de almacenamiento el
producto mantiene su calidad durante su plazo de validez?
|
|
|
|
|
A.5.7.13.1.
¿Se sigue el
procedimiento?
|
|
|
|
|
A.5.7.14.
¿Se llevan
registros de las quejas recibidas sobre la calidad de los productos o
cualquier modificación de sus características físicas así como de las
resoluciones tomadas?
|
|
|
|
|
A.5.7.14.1. ¿Las acciones tomadas con relación
al reclamo y devolución de productos son registradas a fin de saber cuál fue
la actitud tomada?
|
|
|
|
|
A.5.7.15. ¿Existe en la empresa un programa
de validación para los ciclos de esterilización por calor húmedo?
|
|
|
|
|
A.5.7.15.1. ¿Se cumple?
|
|
|
|
|
A.5.7.15.2. ¿Existen protocolos
preestablecidos?
|
|
|
|
|
A.5.7.15.3. ¿Existen registros?
|
|
|
|
|
A.5.7.16.
¿Existe en las
empresas un programa de verificación documentada para métodos analíticos de
control no codificados?
|
|
|
|
|
A.5.7.16.1.
¿Se cumple?
|
|
|
|
|
A.5.7.16.2.
¿Existen protocolos
preestablecidos?
|
|
|
|
|
A.5.7.16.3. ¿Existen registros?
|
|
|
|
|
A.5.7.17. ¿Se realiza una nueva verificación documentada
toda vez que se efectúe un cambio que pueda afectar la calidad o la
reproducibilidad de un proceso o de un método analítico de control?
|
|
|
|
ANEXO B
ESPECIFICACIONES Y CONTROL DE MATERIAS PRIMAS PARA
SOLUCIONES PARENTERALES DE GRAN VOLUMEN
CONTENIDO
B-1 OBJETIVO
B-2 AGUA PARA INYECTABLES
B-3 PRINCIPIOS ACTIVOS (DROGAS)
B-4 REACTIVOS Y METODOS GENERALES
B-1. OBJETIVO
Esta norma establece especificaciones
para las materias primas que deben ser utilizadas en la fabricación de SPGV, y
normaliza los procedimientos respectivos para sus controles.
B-2. AGUA PARA INYECTABLES (RQ)
El agua para inyectables es el agua
obtenida a través de un sistema de producción según las metodologías
establecidas en las ediciones vigentes de la Farmacopea Europea y de la Farmacopea de Estados Unidos de Norteamérica, monitoreado y
validado, que garantice las especificaciones descriptas en las mismas.
Nota: Cuando se la utilice en la
preparación de soluciones parenterales que van a ser esterilizadas
terminalmente, deben tomarse medidas adecuadas para minimizar el crecimiento
bacteriano, o esterilizar previamente el agua para inyectables y luego
protegerla de la contaminación microbiana.
B-2.1. Especificaciones y procedimientos de control
B-2.1.1. pH (USP XXII - 791). Entre 5.0 y 7.0, determinado potenciométricamente
en una solución preparada por la adición de 0.3 mL de solución saturada de
cloruro de potasio por cada 100 mL de la muestra de ensayo.
B-2.1.2. Metales pesados. Tomar 40 mL de agua para
inyectables, ajustar a un pH entre 3.0 y 4.0 con ácido acético 1N, agregar 10
mL de sulfuro de hidrógeno SR recientemente preparado y dejar en reposo durante
10 minutos. Observar sobre fondo blanco: la muestra no debe presentarse más
oscura que 50 mL de la misma agua adicionada de la misma cantidad de ácido
acético 1N (pH entre 3.0 y 4.0), empleándose en la comparación tubos iguales.
B-2.1.3. Calcio. A 100 mL de agua para inyectables
agregarle 2 mL de oxalato de amonio SR: no debe aparecer turbiedad.
B-2.1.4. Amonio. A 100 mL de agua para
inyectables agregarle 2 mL de iodo mercuriato de potasio alcalino SR: el color
amarillento producido inmediatamente no debe ser más oscuro que el de un
control preparado con agua purificada de alta pureza que contenga 0.3 mg/L de
NH3.
B-2.1.5. Cloruro. A 100 mL de agua para
inyectables adicionarle 5 gotas de ácido nítrico y 1 mL de nitrato de plata SR:
no debe presentarse turbidez ni opalescencia.
B-2.1.6. Sulfato. A 100 mL de agua para
inyectables adicionarle 1 mL de cloruro de bario SR: la mezcla debe permanecer
límpida.
B-2.1.7. Sustancias oxidables. A 100 mL de agua
para inyectables adicionarle 10 mL de ácido sulfúrico 2N, calentar a
ebullición, agregar 0.1 mL de permanganato de potasio 0.1N, hervir nuevamente
durante 10 minutos: el color rosa no debe desaparecer completamente.
B-2.1.8. Residuos por evaporación. Evaporar en
baño de María 100 mL de agua para inyectables hasta sequedad en una cápsula
previamente tarada. Desecar el residuo a 105 ºC durante 1 hora: el residuo obtenido deberá ser menor a 1 mg (0.001 %).
B-2.1.9. Dióxido de carbono. A 25 mL de agua
para inyectables colocados en una probeta de 50 mililitros con tapa esmerilada
agregarle 25 mL de hidróxido de calcio SR, tapar la probeta y agitar: la mezcla
debe permanecer límpida.
B-2.1.10. Ensayo de piretógenos. De acuerdo al
"Ensayo de piretógenos" (Anexo L.2) utilizando 10 mL de agua para
inyectables previamente isotomizada por kilo de animal.
B-2.1.11. Endotoxinas bacterianas. De acuerdo
al ensayo de "Endotoxinas bacterianas" (Anexo L.3), el agua para
inyectables no debe contener más de 0.25 UE/mL.
B-2.1.12. Recuento total de microorganismos
aerobios viables. Realizar el ensayo sobre tres muestras consecutivas de 250
mL, recogidas en un mismo punto de muestreo, de acuerdo con la técnica
descripta en: Anexo L.6- Ensayo de contaminación microbiana. Límite: 10 microorganismos por cada 100
mL.
B-3. PRINCIPIOS ACTIVOS (DROGAS)
B-3.1. DEXTROSA (RQ)
C6H12O6.H2o 198.17
D- Glucosa, monohidrato
Anhidra 180.16
Dextrosa es un azúcar obtenido generalmente
por la hidrólisis del almidón. Contiene una molécula de agua de hidratación o
es anhidra.
B-3.1.1. Especificaciones y procedimientos de
control
B-3.1.1.1. Envasado y Conservación. Conservar en
recipientes bien cerrados.
B-3.1.1.2. Rotulado. Rotularla para indicar si
está hidratada o es anhidra.
B-3.1.1.3.Identificación. Agregar algunas gotas
de una solución 1 en 20 a 5 mL de tartrato cúprico alcalino SR caliente: se
forma un precipitado rojo copioso de óxido cuproso.
B-3.1.1.4. Color de la solución. Disolver 25 g en agua y llevar a 50.0 mL de solución: la solución no debe presentar un color más intenso que
una solución preparada mezclando 1.0 mL de cloruro cobaltoso SR; 3.0 mL de
cloruro férrico SR y 2 mL de sulfato cúprico SR con agua hasta alcanzar 10 mL y
diluir 3 mL de esta solución con agua hasta 50 mL. Hacer la comparación
observando las soluciones hacia abajo en sendos tubos para la comparación de
color contra una superficie blanca.
B-3.1.1.5. Rotación específica (USP XXII - 781).
Entre + 52,6º y +53.2º, calculada en base anhidra, determinada en una solución
que contiene 10 g de dextrosa y 0.2 mL de hidróxido de amonio 6N cada 100 mL.
B-3.1.1.6. Acidez. Disolver 5.0 g en 50 mL de agua decarbonatada. Agregar fenoftaleína SR y titular con hidróxido de sodio 0.020N
hasta la aparición de un color rosa definido: se requiere para la
neutralización no más de 0.30 mL.
B-3.1.1.7 Agua, Método III (USP XXII - 921).
Sacar a 105ºC durante 16 horas: la forma hidratada pierde entre 7.5% y 9.5% de
su peso, la forma anhidra pierde no más del 0.5% de su peso.
B-3.1.1.8. Residuo por incineración (USP XXII -
281).No más del 0.1%.
B-3.1.1.9. Cloruro (USP XXII - 221). Una porción
de 2.0 g presenta no más cloruro que el correspondiente a 0,50 mL de ácido
clorhídrico 0.020N (0.018%).
B-3.1.1.10. Sulfato (USP XXII - 221). Una porción
de 2.0 g presenta no más sulfato que el correspondiente a 0.50 mL de ácido
sulfúrico 0.020N (0.025%).
B-3.1.1.11. Arsénico, Método I (USP XXII - 211). 1 ppm.
B-3.1.1.12. Metales pesados (USP XXII - 231).
Disolver 4.0 g en agua hasta alcanzar 25 mL de solución: el límite es 5 ppm.
B-3.1.1.13. Dextrina. Hacer un reflujo de 1 g de dextrosa finamente pulverizada con 20 mL de alcohol: se disuelve completamente.
B-3.1.1.14. Almidón soluble, sulfitos. A una
solución de 1 g en 10 mL de agua agregar 1 gota de iodo de SR: el líquido se
colorea de amarillo.
B-3.2. CLORURO DE SODIO
NaCl 58.44
Cloruro de Sodio
El cloruro de sodio contiene no menos
del 99.0% y no más del 101.0% de NaCl, calculado en base seca. No contiene
sustancias adicionadas.
B-3.2.1. Especificaciones y procedimientos de control
B-3.2.1.1. Envasado y Conservación. Conservar en
recipientes bien cerrados.
B-3.2.1.2. Identificación. Una solución 1 en 20
responde a los ensayos para Sodio y para Cloruro (USP XXII - 191).
B-3.2.1.3. Acidez o alcalinidad. Disolver 50.0 g en 200 mL de agua decarbonatada y agregar 10 gotas del indicador de pH azul de bromotimol SI.
Si la solución es amarilla, requiere no más de 1.0 mL de hidróxido de sodio
0.20N para producir un color azul. Si la solución es azul o verde, requiere no
más de 3.12 mL de ácido clorhídrico 0.20N para producir un color amarillo.
B-3.2.1.4. Pérdida por desecación (USP XXII -
731). Secar a 105 ºC durante 2 horas: pierde no más del 0.5% de su peso.
B-3.2.1.5. Arsénico, Método I (USP XXII - 211). 3 ppm.
B-3.2.1.6. Bario. Disolver 4.0 g en 20 mL de agua, filtrar si es necesario, y dividir la solución en dos porciones. A una
porción, agregar 2 mL de ácido sulfúrico 2N y a la otra agregar 2 mL de agua:
Las soluciones son igualmente claras después de 2 horas de reposo.
B-3.2.1.7. Ioduro o Bromuro. Digerir 2.0 g de cloruro de sodio finamente pulverizado con 25 mL de alcohol caliente durante 3 horas,
enfriar la mezcla y eliminar la sal no disuelta por filtración. Evaporar el
filtrado a sequedad, disolver el residuo en 5 mL de agua, agregar 1 mL de
cloroformo e introducir cuidadosamente, gota a gota, con agitación constante, 5
gotas de cloro SR diluido 1 en 3: el cloroformo no toma color violeta, amarillo
ni naranja.
B-3.2.1.8. Calcio y Magnesio. Disolver 20 g en 200 mL de agua y agregar 0.1 mL de ácido clorhídrico, 5 mL de buffer amonio-cloruro de amonio
SR y 5 gotas de negro de eriocromo SR. Titular con etilendiaminotetraacetato
disódico SV 0.005M hasta punto final azul puro. Cada mL de
etilenediaminotetraacetato disódico 0.005M es equivalente a 0.2004 mg de Ca. Se
encuentra no más del 0.005% de calcio y magnesio (como Ca).
B-3.2.1.9. Hierro (USP XXII - 241). Disolver 5.0 g en 45 mL de agua y 2 mL de ácido clorhídrico: el límite es de 2 ppm.
B-3.2.1.10. Sulfato (USP XXII - 221). Una porción
de 1.0 g presenta no más sulfato que el correspondiente a 0.15 mL de ácido
sulfúrico 0.020N (0.015%).
B-3.2.1.11. Ferrocianuro de sodio. Disolver 25 g en 80 mL de agua en un frasco o probeta graduada de 100 mL con tapón de vidrio. Agregar 2 mL de
sulfato ferroso SR y 1 mL de ácido sulfúrico 2N, diluir con agua a 100 mL y
mezclar. Como control, colocar 80 mL de agua en un frasco o probeta graduada de
100 mL con tapón de vidrio, agregar 2 mL de sulfato ferroso SR y 1 mL de ácido
sulfúrico 2N, diluir con agua a 100 mL y mezclar. Transferir porciones de 50 mL
de las respectivas soluciones a sendos tubos de comparación: la solución de
prueba no debe presentar una coloración azul más intensa que el control,
indicando la ausencia de ferrocianuro de sodio.
B-3.2.1.12. Metales pesados, Método I (USP XXII - 231). 5 ppm.
B-3.2.1.13. Potasio. No más de 500 ppm
determinado por espectrofotometría de llama (Emisión) empleando una solución de
1 % P/V y realizando la medida a 768 mm.
B-3.2.1.14. Valoración. Transferir
aproximadamente 250 mg de cloruro de sodio, exactamente pesados, a una cápsula
de porcelana, y agregar 140 mL de agua y 1 mL de diclorofluoresceína SR.
Mezclar y titular con nitrato de plata
SV 0.1N hasta que flocule el cloruro de plata y la mezcla tome color rosa
claro. Cada mL de nitrato de plata 0.1N es equivalente a 5.844 mg de NaCl.
B-3.3. CLORURO DE POTASIO (RQ)
KCl 74.55
Cloruro de potasio
El cloruro de potasio contiene no
menos del 99.0% y no más del 100.5% de KCl, calculado en base seca.
B-3.3.1. Especificaciones y procedimiento de
control.
B-3.3.1.1. Envasado y Conservación. Conservar en
recipientes bien cerrados.
B-3.3.1.2. Identificación. Una
solución 1 en 20 responde a los ensayos para Potasio y para Cloruro. (USP XXII
- 191).
B-3.3.1.3. Acidez o alcalinidad. A una solución
de 5.0 g en 50 mL de agua decarbonatada agregar 3 gotas de fenoftaleína SR: no
se produce color rosa. Luego agregar 0.30 ml de hidróxido de sodio (0.020N: se
produce un color rosa).
B-3.3.1.4. Pérdida por desecación (USP XXII -
731). Sacar a 105 ºC durante 2 horas: pierde no más del 1% de su peso.
B-3.3.1.5. Ioduro o bromuro. Disolver 2 g en 6 mL de agua, agregar 1 mL de cloroformo y luego agregar, gota a gota, con constante
agitación, 5 mL de una mezcla de partes iguales de cloro SR y agua: el
cloroformo no presenta un color violeta transitorio ni naranja permanente.
B-3.3.1.6. Arsénico, Método 1 (USP XXII - 211). 3 ppm.
B-3.3.1.7. Calcio y magnesio. A 20 mL de una
solución 1 en 100 agregar 2 mL de hidróxido de amonio 6N, 2 mL de oxalato de
amonio SR y 2 mL de fosfato sódico dibásico SR. no se produce turbidez dentro
de los 5 minutos.
B-3.3.1.8. Metales pesados (USP XXII - 231).
Disolver 2.0 g en 25 mL de agua: el límite es 0.001%.
B-3.3.1.9 Sodio. Una solución 1 en 20 ensayada
sobre alambre de platino, no produce un color amarillo pronunciado a la llama
no luminosa.
B-3.3.1.10 Valoración. Pésese exactamente
alrededor de 0.25 gramos de cloruro de potasio previamente desecado, y
transfiérase a un frasco con tapa de vidrio. Agréguense sucesivamente, 50 mL de
solución 0.1N de nitrato de plata, 3 mL de ácido nítrico y 5 mL de
nitrobenceno; mézclese y agítese la mezcla fuertemente, medio a un minuto; añádanse
2 mL de solución de sulfato férrico amónico SR, y valórese el exceso de nitrato
de plata con solución 0.1N de tiocianato de amonio hasta coloración
pardo-rojiza, la cual después de la agitación, no deberá decolorarse al cabo de
cinco minutos.
Cada milímetro de solución 0.1N de
nitrato de plata equivale a 0.0075 gramo de KCL.
B-3.4. ACETATO DE SODIO (RQ)
CH3COONa3H2O
136.08
anhidro 82.03
El acetato de sodio contiene 3
moléculas de agua de hidratación o es anhidro. Contiene un mínimo de 99.0% y un
máximo de 101.0% de C3H3NaO2, calculada en
relación a sustancia seca.
B-3.4.1. Especificaciones y procedimientos de
control.
B-3.4.1.1. Envasado y Conservación. Conservar en
recipientes de cierre perfecto.
B-3.4.1.2. Rotulado. El rótulo debe indicar si se
trata del trihidrato o si es anhidro.
B-3.4.1.3. Identificación. La solución responde a
los ensayos de Sodio y de Acetato (USP XXII - 191).
B-3.4.1.4. pH (USP XXII - 791). Entre 7.5 y 9.2
en una solución 1 en 20 en agua decarbonatada que contenga el equivalente a 30
mg de acetato de sodio anhidro por mL.
B-3.4.1.5. Pérdida por desecación (USP XXII -
731). Secar a 120 ºC hasta peso constante, la forma hidratada pierde entre
38.0% y 41.0% de su peso. La forma anhidra pierde no más del 1% de su peso.
B-3.4.1.6. Material insoluble. Disolver 20 g de acetato de sodio anhidro en 150 mL de agua, calentar a ebullición y digerir durante 1 hora a
baño de María en un recipiente tapado. Filtrar a través de un filtro tarado,
lavar bien y secar a 105 ºC el peso del residuo no excede 10 mg (0.05%).
B-3.4.1.7. Cloruro (USP XXII - 221). Una porción
equivalente a 1.0 g de acetato de sodio anhidro presenta no más cloruro que el
correspondiente a 0.50 mL de ácido clorhídrico 0.020N (0.035%).
B-3.4.1.8. Sulfato (USP XXII - 221). Una porción
equivalente a 10 g de acetato de sodio anhidro presenta no más sulfato que el
correspondiente a 0.50 mL de ácido sulfúrico 0.020N (0.005%).
B-3.4.1.9. Arsénico, Método I (USP XXII - 211). Disolver una porción equivalente a 1.0 g de acetato de sodio anhidro en 35 mL: el límite es 3 ppm.
B-3.4.1.10. Calcio y magnesio. A 20 mL de una
solución que contiene el equivalente a 10 mg de acetato de sodio anhidro por
mL, agregarle 2 mL de hidróxido de amonio 6N, 2 mL de oxalato de amonio SR y 2
mL de fosfato de sodio dibásico SR: no debe presentar turbidez dentro de los 5
minutos.
B-3.4.1.11. Potasio. Disolver el equivalente de 3 g de acetato de sodio anhidro en 5 mL de agua y adicionar 0.2 mL de bitartrato de sodio SR: no debe
presentar turbidez dentro de los 5 minutos.
B-3.4.1.12. Metales pesados, Método I (USP XXII - 231). 0.001% en base seca, se debe usar
ácido acético glacial en lugar de ácido acético diluido para el ajuste del pH.
B-3.4.1.13. Valoración. Pesar exactamente
alrededor de 200 mg de acetato de sodio anhidro y disolver en 25 mL de ácido
acético glacial, calentar suavemente, en caso de ser necesario, hasta
disolución completa. Agregar 2 gotas de p-naftolbenzeína SR y titular con
-ácido perclórico 0.1N SV. Hacer un blanco y efectuar cualquier corrección
necesaria. Cada mL de ácido
perclórico 0.1N SV equivale a 8.203 mg de C2H3NaO2.
B-3. 5. CLORURO DE MAGNESIO (RQ)
MgCl2.6H2O 203.30
Cloruro de Magnesio, hexahidrato
Anhidro 95.21
El Cloruro de Magnesio contiene no
menos del 98.0% y no más del 101.0% de MgCl2.6H2O.
B-3.5.1. Especificaciones y procedimientos de
control.
B-3.5.1.1. Envasado y conservación. Conservar en
recipientes de cierre perfecto.
B-3.5.1.2. Identificación. Una solución 1 en 20
responde a los ensayos para Magnesio y Cloruro (USP XXII - 191).
B-3.5.1.3. pH (USP XXII - 791). Entre 4.5 y 7.0
en una solución 1 en 20 en agua decarbonatada.
B-3.5.1.4. Materia insoluble. Disolver 20 g, exactamente pesados, en 200 mL de agua, calentar a ebullición y digerir en un vaso tapado en un
baño de vapor durante 1 hora. Filtrar a través de un crisol de filtrado tarado,
lavar minuciosamente, y secar a 115 ºC: el peso del residuo no excede 1 mg
(0.005%).
B-3.5.1.5. Sulfato (USP XXII -221). Una porción
de 10 g presenta no más sulfato que el correspondiente a 0.50 mL de ácido
sulfúrico 0.020N (0.005%).
B-3.5.1.6. Arsénico, Método I (USP XXII -211). 3 ppm
B-3.5.1.7. Bario. Disolver 1 g en 10 mL de agua, y agregar 1 mL de ácido sulfúrico 2N: no se produce turbidez dentro de las 2
horas.
B-3.5.1.8. Calcio (USP XXII - 216). No más del
0.01%, usando una muestra de 10 g y usando 0.50 mL de la solución standard de
ión calcio en la preparación standard.
B-3.5.1.9. Potasio. Disolver 5 g en 5 mL de agua, y agregar 0.2 mL de bitrartrato de sodio SR: no se produce turbidez dentro de
los 5 minutos.
B-3.5.1.10. Metales pesados (USP XXII - 231).
Disolver 2 g en agua hasta llegar a 25 mL: el límite es 0.001 %.
B-3.5.1.11. Valoración. Pesar exactamente
aproximadamente 450 mg de cloruro de magnesio, disolver en 25 mL de agua, agregar
5 mL de buffer amonio-cloruro de amonio SR y 0.1 mL de negro de eriocromo SR y
titular con etilendiaminotetraacetato disódico SV 0.05M hasta punto final azul.
Cada mL de
etilendiaminotetraacetato disódico 0.05M es equivalente a 10.17 mg de Mg2Cl26H2O.
B-3.6. CLORURO DE CALCIO (RQ)
CaCl2 .2H2O 147.07
Cloruro de calcio, dihidrato
Anhidro 110.99
El cloruro de calcio contiene una
cantidad de CaCl2 equivalente a no menos del 99.0% y no más del
107.0% de CaCl2.2H2O.
B-3.6.1. Especificaciones y procedimiento de
control
B-3.6.1.1.Envasado y conservación. Conservar en
recipientes de cierre perfecto.
B-3.6.1.2. Identificación. Una solución 1 en 10
responde a los ensayos para Calcio y para Cloruro (USP XXII - 191).
B-3.6.1.3. pH (USP XXII - 791). Entre 4.5 y 9.2,
en una solución 1 en 20.
B-3.6.1.4. Arsénico, Método I (USP XXII - 211). 3 ppm.
B-3.6.1.5. Mezclar pesados (USP XXII-231).
Disolver 2.0 g en 25 mL de agua: el límite es de 0.001%.
B-3. 6. 1. 6 Hierro, aluminio y fosfato. A una
solución 1 en 20 agregar 2 gotas de ácido clorhídrico 3N y una gota de
fenoftaleína SR. Luego agregar cloruro de amonio-hidróxido de amonio SR, gota a
gota, hasta que la solución se torne rosa claro, agregar 2 gotas en exceso, y
calentar el líquido a ebullición: no se produce turbidez o precipitado.
B-3.6.1.7. Magnesio y sales alcalinas. Disolver 1 g en aproximadamente 50 mL de agua, agregar 500 mg de cloruro de amonio, agregar rápidamente 40 mL
de ácido oxálico SR y agitar vigorosamente hasta que se produzca una buena
precipitación. Agregar inmediatamente a la mezcla tibia 2 gotas de rojo de
metilo SR y luego, gota a gota hidróxido de amonio 6N hasta que la mezcla esté
apenas alcalina. Enfriar a temperatura ambiente, transferir a una probeta
graduada de 100 mL, diluir con agua a 100 mL, mezclar y dejar en reposo 4 horas
o toda la noche. Filtrar y agregar a 50 mL del filtrado límpido en una cápsula
de platino 0.5 mL de ácido sulfúrico y evaporar la mezcla hasta pequeño volumen
sobre un baño de vapor. Calentar cuidadosamente sobre llama hasta sequedad y
continuar calentando hasta completa descomposición y volatilización de las
sales de amonio. Finalmente calcinar hasta peso constante: el peso del residuo
no excede los 5 mg (1.0%).
B-3.6.1.8. Valoración. Pesar exactamente
aproximadamente 1 g de cloruro de calcio, transferirlo a un frasco de 250 mL, y
disolverlo en una mezcla de 100 mL de agua y 5 mL de ácido clorhídrico 3N.
Transferir la solución a un frasco volumétrico de 250 mL, diluir con agua hasta
volumen, y mezclar. Pipetear 50 mL de la solución en un recipiente adecuado,
agregar 100 mL de agua, 15 mL de hidróxido de sodio 1N y 300 mg de indicador de
azul de hidroxi-naftol, y titular con etilendiaminotetraacetato disódico SV
0.05M hasta que la solución tenga un color azul profundo. Cada mL de etilendiaminotetraacetato
disódico 0.05M es equivalente a 7.351 mg de CaCl2.2H2O.
B-3.7. CITRATO DE SODIO (RQ)
C6H5Na3O7
(anhidro) 258.07
Sal trisódica del ácido
2-hidrox-1,2,3-Propantricarboxílico
Citrato trisódico (anhidro)
Citrato trisódico dihidrato 294.10
El citrato de sodio es anhidro o
contiene dos moléculas de agua de hidratación. Contiene no menos del 99.0% y no
más del 100,5% de C6H5Na3O7,
calculado en base anhidra.
B-3.7.1. Especificaciones y procedimientos de control.
B-3.7.1.1. Envasado y conservación. Conservar en
recipiente de cierre perfecto.
B-3.7.1.2. Rotulado. Rotularlo para indicar si es
anhidro o está hidratado.
B-3.7.1.3. Identificación.
a) Una solución 1 en 20 responde a los ensayos para
sodio y para citrato (USP XXII - 191).
b) Al incinerar, se obtiene un residuo alcalino que
produce efervescencia cuando es tratado con ácido clorhídrico 3N.
B-3.7.1.4. Alcalinidad. Una solución de 1.0 g en 20 mL de agua es alcalina al papel de tornasol, pero después del agregado de 0.20 mL de
ácido sulfúrico 0.10N no se produce color rosa con 1 gota de fenolftaleína SR.
B-3.7.1.5. Agua, Método III (USP XXII - 921).
Secarlo a 180 ºC durante 18 horas: la forma anhidra pierde no más del 1.0%., y
la forma hidratada, entre 10.0% y el 13.0% de su peso.
B-3.7.1.6. Tartrato. A una solución de 1 g en 20 mL de agua, agregar 1 mL de acetato de potasio SR y 1 mL de ácido acético 6N. Frotar la
pared del tubo con varilla de vidrio: no se forma precipitado cristalino.
B-3.7.1.7. Metales pesados (USP XXII -231). Disolver
2.0 g en 25 mL de agua, y proceder como está indicado en Preparación de
ensayo, excepto usar ácido acético glacial para ajustar el pH: el límite es
0.001 %.
B-3.7.1.8. Valoración. Transferir
aproximadamente 350 mg de citrato de sodio, previamente secados a 180 ºC durante 18 horas y exactamente pesados, a un vaso de 250 mL. Agregar 100 mL de ácido acético
glacial, agitar hasta completa disolución, y titular con ácido perclórico SV
0.1N, determinando el punto final potenciométricamente. Realizar la determinación
de un blanco y hacer cualquier corrección que fuera necesaria. Cada mL de ácido perclórico 0.1N es
equivalente a 8.602 mg de C6H5Na3O7.
B-3.8. GLICERINA (RQ)
C3H8O3
92.09
1,2,3-Propanotriol
Glicerol
La glicerina contiene no menos del
95.0% y no más del 101.0% de C3H8O3.
B-3.8.1. Especificaciones y procedimientos de
control
B-3.8.1.1. Envasado y conservación. Conservar en
recipientes de cierre perfecto.
B-3.8.1.2. Identificación. El espectro de
absorción infrarrojo de una película delgada, muestra una banda muy fuerte y
ancha entre los 2.7 µ
m y 3.3 µ
m, un fuerte doblete a
aproximadamente 3.4 µ
m, un máximo a aproximadamente
6.1 µ
m, una fuerte región de
absorción entre 6.7 µ
m y 8.3 µ
m, teniendo máximas a
aproximadamente 7.1 µ
m, 7.6 µm y 8.2 µm, y una muy
fuerte región de bandas a aproximadamente 9.0 µm, 9.6 µm, 10.1 µm, 10.9 µm y
11.8 µm (Nota: La glicerina que tiene bajo contenido en agua puede no
mostrar un máximo a aproximadamente 6.1 µm).
B-3.8.1.3. Peso específico (USP XXII - 841). No
menos de 1.249.
B-3.8.1.4. Color. Su color, cuando se observa
hacia abajo contra una superficie blanca en un tubo para comparación de color
de 50 mL, no es más oscuro que el color de un standard preparado diluyendo 0.40
mL de cloruro férrico SC con agua hasta 50 mL y observado en forma similar en
un tubo para comparación de color de aproximadamente el mismo diámetro y color
que el que contiene glicerina.
B-3.8.1.5. Residuo por incineración (USP XXII -
281). Calentar 50 g en una cápsula de porcelana de 100 mL abierta, poco
profunda, hasta que se encienda y dejarlos quemar sin más aplicación de calor,
en un lugar sin corrientes de aire. Enfriar, humedecer el residuo con 0.5 mL de
ácido sulfúrico, y calcinar hasta peso constante: el peso del residuo no excede
los 5 mg (0.01 %).
B-3.8.1.6. Cloruro (USP XXII - 221). Una porción
de 7.0 g presenta no más cloruro que el correspondiente a 0.10 mL de ácido
clorhídrico 0.020N (0.001%).
B-3.8.1.7. Sulfato (USP XXII - 221). Una porción
de 10 g presenta no más sulfato que el correspondiente a 0.20 mL de ácido
sulfúrico 0.020N (aproximadamente 0.002%).
B-3.8.1.8. Arsénico. Método I (USP XXII - 211). 1,5 ppm.
B-3.8.1.9. Metales pesados (USP XXII -
231).Mezclar 4.0 g con 2 mL de ácido clorhídrico 0.1N y diluir con agua a 25
mL: el límite es 5 ppm.
B-3.8.1.10. Compuestos clorados. Pesar exactamente
5 g en un balón de 100 mL seco, agregar 15 mL de morfolina, y conectar el
balón mediante una junta esmerilada a un condensador de reflujo. Continuar el
reflujo en forma suave durante 3 horas. Enjuagar el condensador con 10 mL de
agua, recibiendo el lavado en el balón, y acidificando, con precaución, con
ácido nítrico. Transferir la solución a un tubo de comparación adecuado,
agregar 0.50 mL de nitrato de plata 0.10N, diluir con agua hasta 50.0 mL y
mezclar: la turbidez no es mayor que la de un blanco al que se le agregó 0.20
mL de ácido clorhídrico 0.020N, omitiendo el reflujo (0.003% de Cl).
B-3.8.1.11. Acidos grasos y ésteres. Mezclar 50 g con 50 mL de agua recientemente hervida y 5 mL de hidróxido de sodio 0.5N SV, hervir la mezcla 5
minutos, enfriar, agregar fenolftaleína SR, y titular el exceso de álcali con
ácido clorhídrico 0.5N SV. Realizar un blanco: no se consume más de 1 mL de
hidróxido de sodio 0.5N.
B-3.8.1.12. Valoración. Solución de periodato de
sodio: Disolver 60 g de metaperiodato de sodio en agua, que contenga 120 mL de
ácido sulfúrico 0.1N, y llevar a 1000 mL. No calentar para disolver el
periodato. Si la solución no está clara, filtrar a través de un filtro de
vidrio fritado.
Guardar la solución en un recipiente
que lo proteja de la luz, con tapón de vidrio.
Verificar la solución de la siguiente
manera: pipetear 10 mL en un matraz volumétrico de 250 mL, diluir con agua
hasta volumen y mezclar. Agregar con una pipeta 50 mL de la solución de
periodato diluida a unos 550 mg de glicerina disueltos en 50 mL de agua. Como
blanco, pipetear 50 mL de la solución en un matraz que contiene 50 mL de agua.
Dejar descansar las soluciones 30 minutos, luego agregar a cada una 5 mL de
ácido clorhídrico y 10 mL de ioduro de potasio SR, y rotar para mezclar. Dejar
descansar 5 minutos, agregar 100 mL de agua y titular con tiosulfato de sodio
0.1N agitando continuamente y agregando 3 mL de almidón SR a medida que se
acerca el punto final. La relación entre el volumen de tiosulfato de sodio 0.1N
requerido para la mezcla glicerina-periodato respecto del requerido para el
blanco, debería estar entre 0.750 y 0.765.
Procedimiento: Transferir
aproximadamente 400 mg de glicerina, exactamente pesados, a un vaso de 600 mL,
diluir con 50 mL de agua, agregar azul de bromotimol SR, y acidificar con ácido
sulfúrico 0.2N hasta un verde definido o amarillo verdoso. Neutralizar con
hidróxido de sodio 0.05N hasta un punto final azul definido, sin color verde.
Preparar un blanco conteniendo 50 mL de agua, y neutralizar de la misma manera.
Pipetear 50 mL de la solución de
periodato de sodio en cada vaso, mezclar por agitación suave, cubrir con vidrio
de reloj y dejar descansar 30 minutos a temperatura ambiente (no superior a los
35 ºC) en la oscuridad o con luz tenue. Agregar 10 mL de una mezcla de
volúmenes iguales de etilenglicol y agua, dejar descansar 20 minutos.
Diluir cada solución con agua a
aproximadamente 300 mL y titular con hidróxido de sodio 0.1N SV hasta un pH de
8.1 + 0.1 para la muestra en ensayo y 6.5 + 0.1 para el blanco,
usando un pHmetro. Cada mL de hidróxido de sodio 0.1N, después de la corrección
por el blanco, es equivalente a 9.210 mg de C3H8O3.
B-3.9. LACTATO DE SODIO SOLUCION (RQ). La solución de
lactato de sodio es una solución acuosa que contiene no menos del 50.0%, en
peso, de lactato monosódico. Contiene no menos del 98.0% y no más del 102.0% de
la cantidad declarada de C3H5NaO3.
B-3.9.1.Especificaciones y procedimientos de control.
B-3.9.1.1. Envasado y conservación. Conservar en
recipientes de cierre perfecto.
B-3.9.1.2. Rotulado. Rotular la solución para
indicar su contenido en lactato de sodio.
B-3.9.1.3. Identificación. Responde a los
ensayos de Sodio y Lactato (USP XXII - 191).
B-3.9.1.4. pH (USP XXII - 791). Entre 5.0 y 9.0.
B-3. 9.1.5. Cloruro (USP XXII - 221). Una porción,
equivalente a 1 g de lactato de sodio, presenta no más cloruro que el
correspondiente a 0.7 mL de una solución de ácido clorhídrico 0.020N (0.05%).
B-3.9.1.6. Citrato, oxalato, fosfato o tartrato. Diluir 5 mL con agua recientemente
hervida y enfriada, a 50 mL. A 4 mL de esta solución agregar hidróxido de
amonio 6N o ácido clorhídrico 3N, si fuera necesario, para llevar el pH a un
valor entre 7.3 y 7.7. Agregar 1 mL de cloruro de calcio SR, y calentar en un
baño de agua hirviendo durante 5 minutos: la solución permanece clara.
B-3.9.1.7. Sulfato. A 10 mL de una solución 1 en
100 agregar 2 gotas de ácido clorhídrico y 1 mL de cloruro de bario SR: no se
produce turbidez.
B-3.9.1.8. Metales pesados, Método I (USP XXII - 231). Diluir una cantidad de solución
equivalente a 2.0 g de lactato de sodio, con ácido acético 1N hasta 25 mL: el
límite es de 0.001%.
B-3.9.1.9. Azúcares. A 10 mL de tartrato cúprico
alcalino SR caliente agregar 5 gotas de solución: no se forma precipitado rojo.
B-3.9.1.10. Metanol y ésteres metílicos.
Solución de permanganato de potasio y
ácido fosfórico -
Disolver 3 g de permanganato de potasio en una mezcla de 15 mL de ácido
fosfórico y 70 mL de agua. Diluir con agua hasta 100 mL.
Solución de ácido oxálico y ácido
sulfúrico - Agregar,
con precaución, 50 mL de ácido sulfúrico a 50 mL de agua, mezclar, enfriar,
agregar 5 g de ácido oxálico y mezclar hasta disolución.
Preparación standard - Preparar una solución que contenga
10.0 mg de metanol en 100 mL de alcohol diluido 1 en 10.
Preparación de ensayo - Colocar 40.0 g en un balón, con tapón de vidrio, agregar 10 mL de agua, y agregar, con precaución, 30 mL de hidróxido de potasio
5N. Conectar un condensador al balón, y destilar por arrastre con vapor,
recogiendo el destilado en un vaso adecuado de 100 mL, graduado, que contenga
10 mL de alcohol. Continuar la destilación hasta que el volumen en el vaso
receptor alcance aproximadamente 95 mL, y diluir el destilado con agua hasta
100 mL.
Procedimiento - Transferir 10.0 mL de la
preparación standard y de la preparación de ensayo a sendos matraces
volumétricos de 25 mL, agregar a cada uno 5.0 mL de la solución de permanganato
de potasio y ácido fosfórico y mezclar. Después de 15 minutos, agregar a cada
uno, 2.0 mL de la solución de ácido oxálico y ácido sulfúrico, agitar con
varilla de vidrio hasta que la solución quede incolora, agregar 5.0 mL de
fucsina-ácido sulfuroso SR, y diluir con agua hasta volumen. Después de 2
horas, determinar al mismo tiempo las absorbancias de ambas soluciones en
celdas de 1 cm a la longitud de onda de máxima observancia, aproximadamente 575
nm, con un espectrofotómetro apropiado, usando agua como blanco: la absorbancia
de la solución de la preparación de ensayo no debe ser mayor que la de la
preparación standard (0.025%).
B-3.9.1.11. Valoración. Pesar exactamente en un
matraz apropiado, un volumen de la solución de lactato de sodio, equivalente a
aproximadamente 300 mg de lactato de sodio, agregar 60 mL de una mezcla 1 en 5
de anhídrido acético en ácido acético glacial, mezclar, y dejar descansar 20
minutos. Titular con ácido perclórico SV 0.1N, determinando el punto final
potenciométricamente. Realizar la determinación de un blanco, y hacer cualquier
corrección que fuera necesaria. Cada mL de ácido perclórico 0.1N es equivalente a 11.21 mg de C3H5NaO3.
B-3. 10 HIDROXIDO DE SODIO (RQ)
NaOH 40.00
Hidróxido de Sodio
El hidróxido de sodio contiene no
menos del 95.0% y no más del 100.5% de álcali total, calculado como NaOH,
incluyendo no más del 3.0% de Na2CO3.
Precaución: Tener sumo cuidado en el
manipuleo de hidróxido de sodio ya que rápidamente destruye los tejidos.
B-3.10.1. Especificaciones y procedimientos de
control.
B-3.10.1.1. Envasado y conservación. Conservar en
recipientes de cierre perfecto.
B-3.10.1.2. Identificación. Una solución 1 en 25
responde a los ensayos para Sodio (USP XXII - 191).
B-3.10.1.3. Sustancias insolubles y materia
orgánica
Una solución 1 en 20 es total, clara e
incolora a ligeramente coloreada.
B-3.10.1.4. Potasio. Acidificar 5 mL de una
solución 1 en 20 con ácido acético 6N, agregar luego 5 gotas de cobaltinitrito
de sodio SR: no se forma precipitado.
B-3.10.1.5. Metales pesados (USP XXII - 231).
Disolver 0.67 g en una mezcla de 5 mL de agua y 7 mL de ácido clorhídrico 3N.
Calentar hasta ebullición, enfriar y
diluir con agua a 25 mL: el límite es 0.003%.
B-3. 10.1.6. Valoración. Disolver aproximadamente 1.5 g de hidróxido de sodio, exactamente pesados, en aproximadamente 40 mL de agua decarbonatada.
Enfriar la solución a temperatura ambiente, agregar fenolftaleína SR; y titular
con ácido sulfúrico 1N SV.
Al desaparecer el color rosa del
indicador, anotar el volumen de solución ácida requerido, agregar naranja de
metilo SR; y continuar la titulación hasta color rosa persistente. Cada mL de
ácido sulfúrico 1N es equivalente a 40.00 mg de álcali total, calculado como
NaOH, y cada mL de ácido consumido en la titulación con naranja de metilo es
equivalente a 106.0 mg de Na2CO3.
B-3. 11 ACIDO CITRICO (RQ)
C6H8O7
192.12
Acido
2-hidroxi-1,2,3-propantricarboxílico
Acido cítrico
Monohidrato 210.14
El ácido cítrico es anhidro o contiene
una molécula de agua de hidratación. Contiene no menos del 99.5% y no más de
100.5% de C6H8O7, calculado en base anhidra.
B-3.11.1. Especificaciones y procedimientos de
control.
B-3.11.1.1. Envasado y conservación. Conservar en
recipientes de cierre perfecto.
B-3.11.1.2. Rotulado. Rotularlo para indicar si
es anhidro o está hidratado.
B-3.11.1.3. Identificación. Una solución responde
a los ensayos para Citrato (USP XXII - 191).
B-3.11.1.4. Agua, Método I (USP XXII - 921). No
más del 0.5% (forma anhidra) y no más del 8.98% (forma hidratada).
B-3.11.1.5. Residuo por incineración (USP XXII -
281). No más del 0.05%.
B-3.11.1.6. Oxalato. Neutralizar 10 mL de una
solución 1 en 10 con hidróxido de amonio 6N, agregar 5 gotas de ácido
clorhídrico 3N, enfriar, y agregar 2 mL de cloruro de calcio SR: no se produce
turbidez.
B-3.11.1.7. Sulfato. A 10 mL de una solución 1 en
100 agregar 1 mL de cloruro de bario SR al que se le agregó 1 gota de ácido clorhídrico:
no se produce turbidez.
B-3.11.1.8. Arsénico, Método I (USP XXII - 211). 3 ppm.
B-3.11.1.9. Metales pesados (USP XXII - 231).
0.001%.
B-3.11.1.10. Sustancias rápidamente carbonizables.
Transferir 1.0 g, pulverizado para el ensayo, a un tubo de ensayo de 22 x 175 mm previamente enjuagado con 10 mL de ácido sulfúrico SR y dejado escurrir durante 10 minutos.
Agregar 10 mL de ácido sulfúrico SR, agitar hasta disolución completa, y
sumergirlo en un baño de agua a 90 + 1 ºC durante 60 + 0.5 minutos, manteniendo el nivel de ácido por debajo del nivel de agua
durante todo el tiempo. Enfriar el tubo en agua corriente, y transferir el
ácido a un tubo para comparación de color: el color del ácido no es más oscuro
que el de un volumen similar de líquido de comparación de la siguiente
composición: 0.5 partes de cloruro cobaltoso SC y 4.5 partes de cloruro férrico
SC, en un tubo de comparación, observándose los tubos verticalmente contra un
fondo blanco.
B-3.11.1.11. Valoración. Colocar alrededor de 3 g de ácido cítrico en un recipiente tarado, y pesar exactamente. Disolver en 40 mL de agua, agregar
fenolftaleína SR; y titular con hidróxido de sodio IN SV. Cada mL de hidróxido de sodio 1N es
equivalente a 64.04 mg de C6H8O7.
B-3.12. ACIDO LACTICO (RQ)
Acido 2-hidroxi-propanoico
Acido láctico
El ácido láctico es una mezcla de
ácido láctico (C3H6O3) y de lactato de ácido
láctico (C6H10O5) equivalente a un total de no
menos del 85.0% y no más del 90.0%, en peso, de C3H6O3.
Se obtiene por fermentación láctica de azúcares o se prepara sintéticamente. El
ácido láctico obtenido por fermentación de azúcares es levorotatorio, mientras
que el preparado sintéticamente es racémico. (Nota: el ácido láctico preparado
por fermentación se hace dextrorotatorio por dilución, que hidroliza el L(-)
lactato de ácido láctico a L(+) ácido láctico).
B-3.12.1. Especificaciones y procedimientos de
control
B-3.12.1.1. Envasado y conservación. Conservar en
recipientes de cierre perfecto.
B-3. 12. 1.2. Rotulado. Rotularlo para indicar si es
levorotatorio o racémico.
B-3.12.1.3. Identificación. Responde a los
ensayos de lactato (USP XXII - 191).
B-3.12.1.4. Rotación específica (USP XXII - 781).
Entre -0.05º y +0.05º, para ácido láctico racémico.
B-3.12.1.5. Residuo por incineración (USP XXII -
281). No más de 3 mg, de una porción de 5 ml (0.05%).
B-3.12.1.6. Azúcares. A 10 ml de tartrato cúprico
alcalino SR caliente, agregar 5 gotas de ácido láctico: no se forma precipitado
rojo.
B-3.12.1.7. Cloruro. A 10 ml de una solución 1 en
100 acidificada con ácido nítrico, agregar unas pocas gotas de nitrato de plata
SR: no se produce opalescencia inmediatamente.
B-3.12.1.8. Acido cítrico, oxálico, fosfórico o tartárico. A 10 mL de una solución 1 en 10
agregar 40 ml de hidróxido de calcio SR, y hervir durante 2 minutos: no se
produce turbidez.
B-3.12.1.9. Sulfato. A 10 ml de una solución 1 en
100 agregar 2 gotas de ácido clorhídrico y 1 ml de cloruro de bario SR: no se
produce turbidez.
B-3.12.1.10. Metales pesados, Método II (USP XXII
- 231) 0.001%.
B-3.12.1.11. Sustancias rápidamente carbonizables.
Enjuagar un tubo con ácido sulfúrico SR, y dejarlo escurrir durante 10 minutos.
Agregar 5 mL de ácido sulfúrico SR al tubo de ensayo, cubrir cuidadosamente con
5 mL de ácido láctico y, mantener el tubo a una temperatura de 15 ºC; no se desarrolla color oscuro en la interfase de los dos ácidos dentro de los 15 minutos.
B-3.12.1.12. Valoración. A aproximadamente 2.5 mL
de ácido láctico exactamente pesados en un recipiente de 250 mL tarado, agregar
50.0 mL de hidróxido de sodio 1N SV, y hervir la mezcla durante 20 minutos.
Agregar fenoftaleína SR, y titular el exceso de álcali en la solución caliente
con ácido sulfúrico 1N SV. Realizar un blanco. Cada mL de hidróxido de sodio 1N
es equivalente a 90.08 mg de C3H6O3.
B-3. 13 DEXTRAN 40 - (RQ)
Dextrán 40 es un producto obtenido por
descomposición parcial del polisacárido, que es producido por fermentación de
sacarosa con Leuconostoc mesenteroides Van Tieghem (Lactobacillaceae), y su
peso molecular promedio es de aproximadamente 40000.Dextrán 40 se presenta como
un polvo blanco, amorfo e higroscópico. Es inodoro e insípido. Se disuelve
gradualmente en agua. Es completamente soluble en agua caliente, y
prácticamente insoluble en metanol, en etanol y en acetona.
B-3.13.1. Especificaciones y procedimientos de
control.
B-3.13.1.1. Conservación. Conservar en
recipientes de cierre perfecto.
B-3.13.1.2. pH. La solución de dextrán 40 1 en 10
está entre 5.0 y 7.0.
B-3.13.1.3. Identificación. A 1 ml de una solución
de dextrán 40 1 en 3000, agregar 2 ml de antrona SR: aparece un color azul
verdoso, que se torna gradualmente azul verdoso oscuro. Entonces, agregar a
esta solución 1 ml de ácido sulfúrico diluido 1 en 2 ó 1 ml de ácido acético
glacial: la solución no cambia de color.
B-3.13.1.4. Rotación específica. +193.0 - + 201.0º
calculado sobre base anhidra, y determinado en una solución acuosa que contenga
3 g de dextrán 40 en 50 ml.
B-3.13.1.5. Claridad y color de la
solución.Disolver 1.0 g de dextrán 40 en 10 ml de agua por calentamiento: la
solución es incolora y clara.
B-3.13.1.6. Cloruro. Realizar el ensayo con 2.0 g de dextrán 40. Preparar la solución control con 1.0 mL de ácido clorhídrico 0.01N: no más de
0.018 %.
B-3.13.1.7. Metales pesados. Proceder con 1.0 g de dextrán 40 de acuerdo al método I, y realizar el ensayo. Preparar la solución control con
2.0 mL de solución standard de plomo: no más de 20 ppm.
B-3.13.1.8. Nitrógeno. Pesar exactamente
aproximadamente 2 g de dextrán 40, previamente desecado a 105 ºC durante 6 horas, y realizar el ensayo según las indicaciones de Determinación de nitrógeno,
donde se usan 10 mL de ácido sulfúrico para la descomposición, y luego se
agregan 45 mL de una solución de hidróxido de sodio 2 en 5: la cantidad de
nitrógeno no es más que 0.010%.
B-3.13.1.9. Sustancias reductoras. Pesar
exactamente 3.00 g de dextrán 40, previamente desecado a 105 ºC durante 6 horas, disolverlo en agua para lograr exactamente 50 ml, y usar esta solución como
solución de muestra. Separadamente, pesar exactamente 0.450 g de glucosa, previamente desecada a 105 ºC durante 6 horas, disolverla en suficiente agua para
alcanzar 500 ml exactamente y usar esta solución como solución control.
Pipetear 5 ml tanto de la solución de muestra como de la solución control,
agregar agua hasta exactamente 50 ml, respectivamente. Pipetear 5 ml de cada
una de estas soluciones diluidas, agregar 5 ml de cobre alcalino SR,
exactamente medido, y calentar 15 minutos en baño de agua. Enfriar, agregar 1
ml de una solución de ioduro de potasio 1 en 40 y 1.5 ml de ácido sulfúrico
diluido, y titular con tiosulfato de sodio 0.005N (indicador: 2 ml de almidón
SR).
El titulante consumido por la solución
de muestra excede el consumido por la solución control.
B-3.13.1.10. Pérdida por desecación. No más del 5 %
(1 g 105 ºC, 6 horas).
B-3.13.1.11. Residuo por incineración. No más del
0.10% (1 g).
B-3.13.1.12. Viscosidad intrínseca.
B-3.13.1.12.1. Dextrán 40. Pesar exactamente de 0.2 a 0.5 g de dextrán 40, previamente desecado a 105 ºC durante 6 horas, disolver en suficiente agua
hasta exactamente 100 mL, y usar esta solución como solución de muestra.
Realizar el ensayo con la solución de muestra y con agua como se explica en
Viscosidad a 25 + 0.1º: la viscosidad intrínseca, calculada a partir de
la siguiente ecuación, debe estar entre 0.16 y 0.19.
Tiempo de escurrimiento (seg) de la solución muestra
In
-----------------------------------------------------------------
Tiempo
de escurrimiento (seg) del agua
Viscosidad intrínseca =
-----------------------------------------------------------------------------
Cantidad (g) de la muestra
B-3.13.1.12.2. Fracción de alto peso molecular. Pesar
exactamente alrededor de 6 g de dextrán 40, previamente desecado a 105 ºC durante 6 horas, disolver en suficiente agua para alcanzar exactamente 100 mL, y transferir a
un balón. Agregar lentamente suficiente metanol para precipitar del 7 al 10% de
la muestra (usualmente 80 a 90 mL), a 25 + 1 ºC con agitación. Disolver el precipitado a 35 ºC en baño de agua con agitación ocasional y dejarlo
descansar más de 15 horas a 25 + 1 ºC. Separar el sobrenadante líquido por decantación, y calentar el precipitado de la capa inferior a sequedad en
baño de agua. Secar el residuo a 105 ºC durante 6 horas, y calcular la
viscosidad intrínseca del residuo seco según se indica en B-3. 13.1.12.1.: el
valor es no más de 0.27.
B-3.13.1.12.3. Fracción de bajo peso molecular.
Pesar exactamente alrededor de 6 g de dextrán 40, previamente desecado a los 105 ºC durante 6 horas, disolver en suficiente agua hasta alcanzar exactamente 100 mL y transferir a
un balón. Agregar lentamente suficiente metanol para precipitar del 90 al 93%
de la muestra (usualmente 115 a 135 mL) a 25 ± 1 ºC con agitación, centrifugar a 25 ºC, y evaporar el sobrenadante líquido a sequedad en baño de agua.
Desecar el residuo a 105 ºC durante 6 horas, y calcular la viscosidad
intrínseca del residuo seco según se indica en B-3. 13. 1. 12. 1: el valor es
no menos de 0.09.
B-3.13.1.13. Antigenicidad. Disolver 10.0 g de dextrán 40 en solución isotónica de cloruro de sodio para alcanzar 100 mL, y esterilizar.
Con esta solución proceder según se indica en antigenicidad en dextrán 40
inyectable.
B-3.13.1.14 Piretógenos. Disolver 10.0 g de dextrán 40 en solución isotónica de cloruro de sodio hasta 100 mL, y realizar el ensayo:
esta solución cumple los requerimientos del ensayo de piretógenos.
B-3.14. DEXTRAN 70 (RQ)
Dextrán 70 es un producto obtenido por
descomposición parcial del polisacárido, que es producido por fermentación de
sacarosa con Leuconostoc mesenteroides Van Tieghem (Lactobacillaceae), y su
peso molecular promedio es aproximadamente 70000.
Dextrán 70 se presenta como un polvo
blanco, amorfo e higroscópico. Es inodoro e insípido. Se disuelve gradualmente
en agua. Es totalmente soluble en agua caliente, y prácticamente insoluble en
metanol, en etanol y en acetona.
B-3.14.1. Especificaciones y procedimientos de
control
B-3.14.1.1. Conservación. Conservar en
recipientes de cierre perfecto.
B-3.14.1.2. pH. La solución de dextrán 70 3 en 50
está entre 5.0 y 7.0.
B-3.14.1.3. Identificación. Proceder como se
indica en la identificación de dextrán 40.
B-3.14.1.4. Rotación específica +193.0 - +201.0º
calculado sobre base anhidra, y determinado en una solución que contenga 3 g de dextrán 70 en 50 mL.
B-3.14.1.5. Claridad y color de la solución.
Disolver 1.0 g de dextrán 70 en 10 mL de agua con calor: la solución es
incolora y clara.
B-3.14.1.6. Cloruro. Realizar el ensayo con 2.0 g de dextrán 70. Preparar la solución control con 1.0 mL de ácido clorhídrico 0.01N: no más de
0.018%.
B-3.14.1.7. Metales pesados. Proceder con 1.0 g de dextrán 70 de acuerdo al Método I y realizar el ensayo. Preparar la solución control con 2.0
mL de solución standard de plomo: no más de 20 ppm.
B-3.14.1.8. Nitrógeno. Pesar exactamente aproximadamente
2 g de dextrán 70, previamente desecado a 105 ºC durante 6 horas, y realizar el ensayo según las indicaciones de Determinación de Nitrógeno,
donde se usan 10 mL de ácido sulfúrico para la descomposición, y se agregan 45
mL de una solución de hidróxido de sodio 2 en 5: la cantidad de nitrógeno no es
más que 0.010%.
B-3.14.1.9. Sustancias reductoras. Pesar
exactamente 3.00 g de dextrán 70, previamente desecado a 105 ºC durante 6 horas, disolverlo en agua para lograr exactamente 50 mL, y usar esta solución como
solución de muestra. Separadamente, pesar exactamente 0.300 g de glucosa, previamente desecada a 105 ºC durante 6 horas, disolverla en suficiente agua para
alcanzar 500 mL exactamente y usar esta solución como solución control.
Pipetear 5 mL tanto de la solución de muestra como de la solución control, y
agregar agua hasta exactamente 50 mL, respectivamente. Pipetear 5 mL de cada
una de estas soluciones diluidas, agregar 5 mL de cobre alcalino SR,
exactamente medido, y calentar 15 minutos en baño de agua. Enfriar, agregar 1
mL de una solución de ioduro de potasio 1 en 40 y 1.5 mL de ácido sulfúrico
diluido, titular con tiosulfato de sodio 0.005N (indicador: 2 mL de almidón
SR).
El titulante consumido por la solución
de muestra excede el consumido por la solución control.
B-3.14.1.10 Pérdida por desecación. No mas del
5.0% (1 g, 105 ºC, 6 horas)
B-3.14.1.11. Residuo por incineración. No más de
0.10% (1 g).
B-3.14.1.12. Viscosidad intrínseca
B-3.14.1.12. 1 Dextrán 70. Pesar exactamente de 0.2 a 0.5 g de dextrán 70, previamente desecado a los 105 ºC durante 6 horas, disolverlo en
suficiente agua hasta exactamente 100 mL, y usar esta solución como solución
muestra. Realizar el ensayo con la solución de muestra y con agua como se
explica en viscosidad a 25 + 0.1 ºC: la viscosidad intrínseca, calculada
a partir de la siguiente ecuación, debe estar entre 0.21 y 0.26.
tiempo de escurrimiento (seg) de la solución muestra
In ----------------------------------------------------------------------------
Tiempo de escurrimiento (seg) del agua
Viscosidad intrínseca =
--------------------------------------------------------------------------
Cantidad (g) de la muestra
B-3.14.1.12.2. Fracción de alto peso molecular. Pesar
exactamente alrededor de 6 g de dextrán 70, previamente desecado a 105 ºC durante 6 horas, disolver en suficiente agua para alcanzar exactamente 100 mL, y transferir a
un balón. Agregar lentamente suficiente metanol para precipitar del 7% al 10%
de la muestra (usualmente 75 a 85 mL), a 25 + 1 ºC con agitación. Disolver el precipitado en baño de agua a 35 ºC con agitación ocasional y dejarlo descansar más de 15 horas a 25 + 1 ºC. Separar el sobrenadante líquido por decantación y calentar el precipitado de la capa inferior a sequedad en baño
de agua. Secar el residuo a 105 ºC durante 6 horas, y calcular la viscosidad
intrínseca del residuo seco según se indica en 3. 14. 1. 12. 1: el valor es no
más de 0.35.
B-3.14.1.12.3. Fracción de bajo peso molecular. Pesar
exactamente alrededor de 6 g de dextrán 70, previamente desecado a los 105 ºC durante 6 horas, disolver en suficiente agua hasta alcanzar exactamente 100 mL, y transferir a
un balón. Agregar lentamente suficiente metanol para precipitar del 90 al 93%
de la muestra (usualmente 110 a 130 mL) a 25 + 1 ºC con agitación, centrifugar a 25 ºC, y evaporar el sobrenadante líquido a sequedad en baño de
agua. Desecar el residuo a 105 ºC durante 6 horas, y calcular la viscosidad
intrínseca del residuo seco según se indica en 3. 14. 1. 12. 1: el valor es no
menos de 0.10.
B-3.14.1.13. Antigenicidad. Disolver 6.0 g de dextrán 70 en solución isotónica de cloruro de sodio para alcanzar 100 mL, y esterilizar.
Con esta solución, proceder según se indica en antigenicidad en dextrán 40
inyectable.
B-3.14.1.14. Piretógenos. Disolver 6.0 g de dextrán 70 en solución isotónica de cloruro de sodio hasta 100 mL. Realizar el ensayo: esta
solución cumple los requerimientos del ensayo de piretógenos.
B-3.15. MANITOL (RQ)
C6H14O6
182.17
Manita
El Manitol es un alcohol
hexahidroxilado derivado de la manosa que contiene no menos de 98 por ciento de
C6H14O6, calculado para la sustancia desecada.
Es un polvo cristalino, blanco, inocuo
y con sabor dulzaíno. Soluble en 6 partes de agua destilada; poco soluble en
alcohol; insoluble en éter.
B-3.15.1. Especificaciones y procedimientos de
control
B-3.15.1.1. Conservación. Conservar en recipientes
de cierre perfecto.
B-3.15.1.2. Identificación
a) En un tubo de ensayo que contiene 1 mililitro de
solución saturada de manitol, añádanse 5 gotas de solución de cloruro férrico
SR. En otro tubo de ensayo que contiene 1 mL de solución de cloruro férrico SR,
añádanse 5 gotas de agua destilada. Añádanse a cada tubo, cinco gotas de
solución concentrada de hidróxido de sodio SR: en el tubo sin manitol deberá
formarse un precipitado pardo de hidróxido de hierro, mientras que en el tubo
que contiene manitol se formará un precipitado amarillo. Agítense los tubos
fuertemente; deberá obtenerse una solución límpida en el tubo con manitol, en
tanto que el precipitado persistirá en el otro tubo. La adición ulterior de solución
concentrada de hidróxilo de sodio SR, no deberá producir precipitación en el
tuvo con manitol, pero sí una nueva precipitación en el otro tubo.
b) En un tubo de ensayo colóquese alrededor de 0.5 g de manitol y agréguense 3 mL de anhídrido acético y 1 mL de piridina. Caliéntese la mezcla en
un baño de María durante 15 minutos o hasta que se haya disuelto completamente,
y caliéntese durante 5 minutos más. Enfríese; agréguense 20 mL de agua
destilada, mézclese y déjese en reposo durante 5 minutos. Recójase el
precipitado en un crisol de vidrio con placa filtrante de vidrio poroso; el
hexaacetato de manitol, así obtenido, después de secado con vacío a 60 ºC durante 1 hora, deberá fundir entre 119 y 124 ºC.
B-3.15.1.3. Punto de fusión. Entre 165 y 168 ºC ablandándose a temperatura más baja.
B-3.15.1.4. Acidez. Disuélvanse 5 g de manitol. en 50 mL de agua destilada exenta de anhídrido carbónico: el líquido obtenido no
deberá requerir para su neutralización más de 0.3 mL de solución 0.02N de
hidróxido de sodio, usando solución de fenoftaleína SR como indicador.
B-3.15.1.5. Rotación específica. El poder
rotatorio específico a 20 ºC de una solución preparada agregando a 5 g de manitol 6.4 g de borato de sodio y cantidad suficiente de agua destilada hasta completar 45
mL; dejando en reposo durante 1 hora, agitando de cuando en cuando, y
completando después hasta 50 mL con agua destilada; no deberá ser menor de +23º
ni mayor de +24º.
B-3.15.1.6. Cloruro. Una porción de 5 g de manitol deberá cumplir el ensayo para límite de cloruros.
B-3.15.1.7. Sulfato. Una porción de 5 g de manitol deberá cumplir el ensayo para límite de sulfatos.
B-3.15.1.8. Azúcares reductores. Disuélvase 0.2 g de manitol, en 2 mL de agua destilada; agréguense 5 mL de solución cupritartárica alcalina SR y
caliéntese en baño de María durante 5 minutos: a lo sumo, no deberá formarse
más que un precipitado muy escaso.
B-3.15.1.9. Pérdida por desecación. Por
desecación a 105 ºC durante 4 horas, no deberá perder más de 0.3% de su peso.
B-3.15.1.10. Residuo por incineración. No deberá
dejar más de 0.1% de residuo.
B-3.15.1.11. Arsénico Método II (USP XXII - 211). 2 ppm.
B-3.15.1.12. Valoración. Pésese exactamente
alrededor de 0.2 g de manitol previamente desecado y transfiérase a un matraz
aforado de 250 mL; disuélvase en agua destilada y dilúyase con este disolvente
hasta completar el volumen. Transfiéranse 5 mL de esa solución a otro matraz
aforado de 250 mL y agréguense 50 mL del siguiente reactivo: mézclense 40 mL de
ácido sulfúrico diluido 1 en 20 con 60 mL de solución de periodato de potasio 1
en 1000 acidificada con 3 a 5 gotas de ácido sulfúrico: Caliéntese la solución
a baño de María durante 15 minutos; enfríese a temperatura ambiente y agréguese
1 g de ioduro de potasio. Déjese en reposo durante 5 minutos y valórese con
solución 0.02N de tiosulfato de sodio, añadiendo solución de almidón SR en la
proximidad del punto final. Repítase la valoración omitiendo el manitol: la
diferencia entre las dos valoraciones representa la cantidad de solución 0.02N de
tiosulfato de sodio requerida por el manitol.
Cada mL de solución 0.02N de
tiosulfato de sodio equivale a 0.00036 g de C6H14O6.
B-3. 16 BICARBONATO DE SODIO (RQ)
CO3HNa 84.01
Carbonato ácido de sodio
Carbonato monosódico
El bicarbonato de sodio contiene no
menos de 99% de CO3HNa, calculado para la sustancia desecada.
Es un polvo blanco o pequeños
cristales opacos, monoclínicos; inodoros; con sabor salino y francamente
alcalino.
Estable en el aire seco, pero en el
aire húmedo pierde poco a poco anhídrido carbónico, transformándose en
carbonato neutro hidratado.
Soluble en 11 partes de agua
destilada; insoluble en alcohol.
B-3.16.1. Especificaciones y procedimientos de
control.
B-3.16.1.1. Conservación. Conservar en recipientes
de cierre perfecto.
B-3.16.1.2. Identificación. La solución de
bicarbonato de sodio deberá responder a los ensayos para bicarbonato y para
sodio.
B-3.16.1.3. pH. El pH de una solución bicarbonato
de sodio al 1% P/V recientemente preparada, no deberá ser mayor de 8.6.
B-3.16.1.4. Aluminio, calcio y materias
insolubles. Hiérvanse 10 g de bicarbonato de sodio con 50 mL de agua destilada
y 20 mL de amoníaco diluido SR; fíltrese; séquese y calcínese el residuo
insoluble: el residuo obtenido no deberá pesar más de 0.001 gramo.
B-3.16.1.5. Carbonato. Disuélvanse 1 g de bicarbonato de sodio, sin agitar, en 20 mL de agua destilada a una temperatura no mayor de 15 ºC, y agréguense 2 mL de solución 0,1N de ácido clorhídrico y dos gotas de solución de
fenolftaleína SR: la mezcla no deberá tomar inmediatamente un tinte rosado.
B-3.16.1.6. Potasio. Disuélvanse 2 g de bicarbonato de sodio en unos 15 mL de agua destilada; agréguense 3 mL de ácido clorhídrico y
evapórese la mezcla hasta la sequedad; caliéntese el residuo a 120 ºC durante media hora y disuélvase en 10 mL de agua destilada. A 5 mL de esta solución,
agréguense 5 mL de solución de nitrito cobáltico sódico SR y 10 mL de alcohol:
de producirse una turbiedad dentro de la media hora, no deberá exceder a la
producida en una solución testigo que contiene el residuo de evaporación de 0.1
mg de potasio y 3 mL de ácido clorhídrico.
B-3.16.1.7. Cloruro. Una porción de 2.5 g de bicarbonato de sodio, disuelta en agua destilada adicionada de 2 mL de ácido nítrico, deberá
cumplir el ensayo para límite de cloruros.
B-3.16.1.8. Sulfato. Una porción de 1 g de bicarbonato de sodio, disuelta en agua destilada adicionada de 6 mL de ácido clorhídrico
diluido SR, deberá cumplir el ensayo para límite de sulfatos.
B-3.16.1.9 .Hierro. Una porción de 2.5 g de bicarbonato de sodio disuelta en una mezcla de 20 mL de agua destilada y 4 mL de ácido
clorhídrico, y diluida con agua destilada hasta completar 40 mL, deberá cumplir
el ensayo para límite de hierro.
B-3.16.1.10. Sales de amonio. Caliéntese 1 g de bicarbonato de sodio con 10 mL de solución de hidróxido de sodio SR: no deberán desprenderse
vapores de amoníaco.
B-3.16.1.11. Metales pesados. Mézclese 2 g de bicarbonato de sodio con 5 mL de agua destilada y 9.5 mL de ácido clorhídrico diluido SR;
hiérvase en baño de María durante 1 minuto; añádase una gota de solución de
fenolftaleína SR y agréguese , gota a gota, amoníaco diluido SR hasta que la
solución adquiera un tinte ligeramente rosado; enfríese; agréguense 2 mL de
ácido acético diluido SR y cantidad suficiente de agua destilada hasta
completar 25 mL: el límite de metales pesados, utilizando un testigo preparado
con la solución tipo de plomo, es 5 ppm.
B-3.16.1.12. Pérdida por desecación. Por
desecación durante 4 horas, en presencia de gel de sílice, una porción de unos 4 g de bicarbonato de sodio exactamente pesada, no deberá perder más de 0.25% de su peso.
B-3.16.1.13. Arsénico Método I (USP XXII - 211). Límite 2 ppm.
B-3.16.1.14. Valoración. Pésense exactamente
alrededor de 3 g de bicarbonato de sodio previamente desecado; disuélvanse en
25 mL de agua destilada y valórese con solución 1N de ácido sulfúrico, usando
solución de anaranjado de metilo SR como indicador.
Cada mililitro de solución 1N de ácido
sulfúrico equivale a 0.0840 gramo de CO3HNa.
B-3.17. SORBITOL (RQ)
C6H14O6
182.17
D-Clucitol
El sorbitol contiene no menos del 91%
y no más del 100.5% de C6H14O6, calculado en
base seca. Puede contener pequeñas cantidades de otros alcoholes polihídricos.
B-3.17.1. Especificaciones y procedimiento de
control.
B-3.17.1.1. Envasado y conservación. Conservar en
recipientes bien cerrados.
B-3.17.1.2. Identificación. Una solución de 5 g en alrededor de 4 mL responde al siguiente ensayo: Tomar 6 mL y agregarle 6 mL de metanal, 1 mL
de benzaldehído y 1 mL de ácido clorhídrico y agitar mecánicamente hasta la
aparición de cristales. Filtrar con vacío, disolver los cristales en 20 mL de
agua en ebullición conteniendo 1 g de bicarbonato de sodio. Filtrar en
caliente, enfriar el filtrado, filtrar con vacío, lavar con 5 mL de una mezcla
de partes iguales de metanol y agua y secar con aire. El derivado
monobencilidensorbitol así obtenido funde a 174-179ºC.
B-3.17.1.3. Agua, Método I (USP XXII - 921). No
más de 1.0%.
B-3.17.1.4. Residuo por incineración (USP XXII -
281). No más de 0.1%.
B-3.17.1.5. Cloruro (USP XXII-221). Una porción
de 1.5 g presenta no más cloruro que el correspondiente a 0.10 mL de ácido
clorhídrico 0.02N (0.0050%).
B-3.17.1.6. Sulfato (USP XXII - 221). Una porción
de 1.0 g presenta no más sulfato que el correspondiente a 0.10 mL de ácido
sulfúrico 0.0020N (0.0025%).
B-3.17.1.7. Arsénico, Método II (USP XXII - 211).
Límite 3 ppm.
B-3.17.1.8. Metales pesados (USP XXII - 231).
Disolver 2.0 g en 25 mL de agua: el límite es 0.001%.
B-3.17.1.9. Azúcares reductores. Transferir 7 g, exactamente pesados, a un vaso de 400 mL con la ayuda de 35 mL de agua y mezclar. Agregar 50 mL
de tartrato cúprico alcalino SR, cubrir el vaso, calentar la mezcla a una
velocidad tal que comience a hervir en 4 minutos y hervir por 2 minutos, midiendo
el tiempo exactamente. Recoger el óxido cuproso precipitado en un crisol
filtrante tarado, previamente lavado con agua caliente, alcohol y éter y luego
secado a 105 ºC por 30 minutos.
Lavar el óxido cuproso en el filtro
con agua caliente, luego con 10 mL de alcohol, y finalmente con 10 mL de éter,
y secar a 105 ºC durante 30 minutos: el peso de óxido cuproso no debe exceder
los 50 mg.
B-3.17.1.10. Valoración. Fase móvil: usar
agua degasificada.
Solución de resolución: disolver Manitol y Sorbitol SR en agua
para obtener una solución de concentración 4,8 mg/mL de cada uno.
Solución standard: disolver una cantidad de Sorbitol SR pesada con
precisión en agua, diluir cuantitativamente con agua para obtener una solución
de concentración conocida de alrededor de 4.8 mg/mL.
Solución de ensayo: transferir alrededor de 0.24 g de Sorbitol, pesados con precisión a un matraz aforado de 50 mL, disolver en 10 mL de agua,
llevar a volumen y mezclar.
Sistema cromatográfico: el cromatógrafo líquido está
equipado con detector de índice de refracción que se mantiene a temperatura
constante, y una columna de 7.8 mm x 30 cm que contiene resina de intercambio catiónico fuerte, consistente en un copolímero de estireno divinilbenceno
entrecruzada, sulfonada, en la forma cálcica y de alrededor de 9 Om de
diámetro.
La columna se mantiene a 30 +/- 2ºC y el flujo es de alrededor de 0.2 mL/minuto. Realizar la cromatografía de la solución standard y
registrar las respectivas respuestas de los picos como se indica en
Procedimiento. La desviación standard relativa para inyecciones replicadas es
no mayor de 2%. De igual manera, realizar la cromatografía de la Solución de Resolución: la resolución, R, entre los picos de sorbitol y manitol es no menor
que 2.0.
Procedimiento: inyectar separadamente volúmenes
iguales (alrededor de 20 Ol) de solución de ensayo y solución estandar, correr
los cromatogramas, medir las respuestas de los picos mayoritarios. Calcular la
cantidad, en mg., de C6H14O6, en el Sorbitol
con la fórmula:
50 c(ru / rs)
donde c es la concentración, en mg/mL,
de Sorbitol RS en la preparación standard, y ru y rs son las respuestas de los
picos obtenidas de la Solución de ensayo y la Solución Standard respectivamente.
B-3. 18 GELATINA (RQ). Gelatina es un producto obtenido
por hidrólisis parcial de colágeno, derivado de la piel, tejido conectivo
blando y huesos de animales. La obtenida de un precursor por tratamiento ácido
es denominada como tipo A, y por tratamiento alcalino tipo B.
B-3.18.1. Especificaciones y procedimiento de
control.
B-3.18.1.1. Envasado y conservación. Conservar en
recipientes bien cerrados y en lugar seco.
B-3.18.1.2 .Identificación
A: Agregar a una solución (1 en 100) trinitrofenol SR
o una solución de dicromato de potasio (1 en 15) previamente mezclada con un cuarto
de su volumen con HCl 3 N: se forma un precipitado amarillo.
B: Agregar a una solución (1 en 5000) ácido tánico SR:
se produce turbidez.
B-3.18.1.3. Límites microbianos. Recuento de
microorganismos aeróbicos totales viables menor a 103 por gramo, Ausencia
de salmonella y Escherichia coli en 10 gramos. (Ensayos de contaminación microbiana Anexo L.6).
B-3.18.1.4. Residuo por incineración (USP XXII -
281). Incinerar 5.0 g sin el uso de ácido sulfúrico, con la adición de 1.5 a 2.0 g de parafina para evitar pérdidas debido a hinchamiento. Luego llevar a cenizas en una
mufla a 550ºC por 15 a 20 horas: el peso del residuo no debe exceder 2.0%.
B-3.18.1.5. Olor y sustancias insolubles en agua.
Una solución caliente (1 en 40) está libre de cualquier olor desagradable y
cuando se observa una capa de 2 cm de espesor es sólo levemente opalescente.
B-3.18.1.6 Dióxido de azufre. Disolver 20.0 g en 150 mL de agua caliente en un balón de cuello alargado, agregar 5 mL de ácido fosfórico y 1 g de bicarbonato de sodio e inmediatamente conectarle un refrigerante (Nota: la formación excesiva
de espuma se puede disminuir con unas pocas gotas de antiespumante).
Destilar 50 mL, recibiendo el
destilado en 50 mL de iodo 0.1N. Acidificar el destilado con unas pocas gotas
de ácido clorhídrico, agregar 2 mL de cloruro de bario SR, y calentar en baño
María hasta que el líquido sea prácticamente incoloro. El precipitado de
sulfato de bario, si lo hubiera, una vez filtrado, lavado e incinerado, pesa no
más de 3 mg correspondiente a no más de 0.004% de dióxido de azufre. Deben
hacerse correcciones por el sulfato que pudiera estar presente en 50 mL de iodo
0.1N.
B-3.18.1.7 Arsénico, Método I (USP XXII - 211). Preparar la solución de ensayo como se
indica a continuación. Mezclar 3.75 g con 10 mL de agua en el generador.
Agregar 10 mL de ácido nítrico y 10 mL de ácido perclórico, mezclar, calentar
cuidadosamente hasta la producción de humos intensos de ácido perclórico.
Enfriar, lavar hacia abajo los laterales del generador con agua, agregar 10 mL
de ácido nítrico y nuevamente calentar hasta humos intensos. Enfriar, lavar y
nuevamente calentar hasta humos intensos. Si es necesario repetir la digestión,
hasta obtener una solución clara. Enfriar, diluir con agua a 52 mL, agregar 3
mL de ácido clorhídrico: la solución resultante, sigue los requerimientos del
ensayo, la adición de 20 mL de ácido sulfúrico 7N especificada en el
procedimiento se omite.
El límite es 0.8 ppm.
B-3.18.1.8. Metales pesados (USP XXII - 231). Al
residuo obtenido en el ensayo B-3. 18. 1. 4 agregarle 2 mL de ácido clorhídrico
y 0.,5 mL de ácido nítrico y evaporar en baño María a sequedad. Agregarle el
residuo 1 mL de ácido clorhídrico 1N y 15 mL de agua y calentar por unos
minutos. Filtrar, lavar con agua hasta obtener 100 mL de filtrado. Diluir 8 mL
de solución a 25 mL con agua.
El límite es 0.005%.
B-4 REACTIVOS Y METODOS GENERALES
B-4.1. Soluciones SR. Ver características, preparación y
usos en: "Test Solutions (TS)" en USP XXII páginas 1786-1792.
B-4.2. Soluciones SV. Ver características preparación y
usos en: "Volumetric Solutions (VS)" en USP XXII páginas 1792-1798.
B-4.3. Métodos generales. Ver metodologías en:
"General Requeriments for Tests and Assays" en USP XXII páginas
1470-1623.
ANEXO C
RECIPIENTES DE VIDRIO PARA SOLUCIONES
PARENTERALES DE GRAN VOLUMEN
CONTENIDO
C-1 OBJETIVO
C-2 DEFINICIONES
C-3 CONDICIONES GENERALES
C-4 CONDICIONES ESPECÍFICAS
C-5 CRITERIOS PARA LA ACEPTACIÓN O RECHAZO
C-1. OBJETIVO
Esta norma fija las condiciones relativas a los
aspectos físicos y químicos de los recipientes de vidrio indicados
específicamente para el envasado de Soluciones Parenterales de Gran Volumen
(SPGV).
C-2. DEFINICIONES
Para los efectos de esta norma se adoptan las
definiciones C. 2.1 y C. 2.2
C-2.1. Vidrio Tipo I. Vidrio borosilicato neutro,
destinado a envasar medicamentos de uso parenteral.
C-2.2. Vidrio Tipo II
- Vidrio sódico-cálcico tratado,
generalmente usado para soluciones parenterales neutras o ácidas. Si se
comprueba su estabilidad en relación a la solución envasada, podrá ser
utilizado para preparaciones parenterales alcalinas.
C-3. CONDICIONES GENERALES
C-3.1. De los frascos. Los frascos deben ser
acondicionados por el fabricante de la siguiente forma: a) dispuesto en forma
ordenada y de boca hacia abajo, b) separados por paredes divisorias, c) cada
ambalaje debe contener un único producto y un único número de lote.
C-3.2. De los embalajes
Los embalajes que acondicionan los
frascos deben ofrecer seguridad durante su transporte y almacenamiento, no
permitir la entrada de contaminantes y obedecer a las siguientes
recomendaciones:
a) estar en buen estado de conservación, es decir, sin
deformaciones, rayaduras, manchas, humedad, o cuerpos extraños.
b) poseer resistencia suficiente para permitir la
estiba y almacenamiento sin sufrir deformaciones.
c) estar identificados, individual y externamente con
la siguiente información:
- identificación del recipiente y tipo
de vidrio
- nombre del producto al que el frasco
se destina (frasco impreso)
- volumen nominal
- cantidad de recipientes por embalaje
- poseer indicación de posicionamiento
(flecha o símbolo)
- poseer indicación de altura máxima
de estiba
- número de lote
- fecha de fabricación
- nombre del fabricante
- código del recipiente (si es
necesario).
C-4. CONDICIONES ESPECIFICAS
Los frascos de vidrio deben poseer las
características y responder a los ensayos descriptos en C-4.1. a C-4.3. Los
ensayos de C-4.1. y C-4.2. se realizarán en muestras tomadas por planes
estadísticos de muestreo.
C-4.1. Inspección visual. Los frascos no deben presentar
los siguientes defectos:
C-4.1.1. Defectos críticos (RQ)
a) texto impreso ilegible o con error de impresión.
b) escala volumétrica ilegible o errónea.
c) fragmentos o rebabas de vidrio adheridas o no a sus
paredes (interna o externa).
d) contaminación por hongos en su interior.
e) manchas de la pared externa o interna,
irremovibles en las condiciones normales de limpieza.
f) boca defectuosa (ovalización, estrangulamiento)
que comprometa la hermeticidad del cierre.
C-4.1.2. Defectos mayores
a) irregularidades de espesor
b) deformación en el cuerpo o en el fondo del frasco
c) inclusión de material no fundido
d) fisura, rayadura o poro en las paredes del frasco
e) burbujas de aire (mayores de 2 mm) en su pared
f) cuello torcido, deformado o incompleto
C-4.2. Ensayos físicos (RQ)
C-4.2.1. Dimensiones: según las especificaciones del diseño
patrón.
C-4.2.2. Volumen: dentro de un límite de tolerancia de +
2% del declarado.
C-4.2.3. Choque térmico. Los frascos destinados a contener
SPGV deben soportar una temperatura diferencial mínima de 42ºC, según el procedimiento siguiente:
Sumergir completamente los frascos en
un baño de agua a 20.0 + 1.5ºC, manteniéndolos hasta que la temperatura
se estabilice.
Transferirlos inmediatamente a un baño
de agua caliente a 65.0 + 1.5ºC, manteniéndolos completamente sumergidos
durante 300 + 10 seg. Sacarlos y volverlos a colocar en el baño de agua
fría durante más de 15 segundos y menos de 1 minuto.
La capacidad de cada baño deberá ser
como mínimo 4.2 L por cada 500 g de vidrio a ser ensayado.
C-4.2.4. Peso: dentro de las tolerancias establecidas.
C-4.3. Ensayos químicos (RQ)
C-4.3.1. Ensayos de resistencia química o hidrolítica.
Determinan la resistencia del material, ya sea sobre el vidrio en polvo o sobre
la superficie interna del recipiente de vidrio, al ataque por agua. El grado de
ataque está determinado por la alcalinidad liberada hacia el medio en las
condiciones especificadas y es extremadamente pequeño en los vidrios de mayor
resistencia.Los ensayos deben ser realizados en ambiente exento de humo y
polvo.
C-4.3.1.1. Ensayo sobre vidrio en polvo, para
vidrio Tipo I. Lavar cuidadosamente con agua para inyectables (conductividad no
mayor de 0,15 µmho/cm a 25ºC) 6 (seis) o más contenedores seleccionados al azar
siguiendo un procedimiento de muestreo convenido.
Secarlos con una corriente de aire
limpio y seco. Romper los contenedores en fragmentos de aproximadamente 25 mm. Dividir aproximadamente 100 g del vidrio así roto en tres porciones aproximadamente iguales,
colocar una de esas porciones en el mortero especial, y moler el vidrio
golpeando 3 ó 4 veces con el martillo. Preparar los tamices de acero inoxidable
de 20.3 cm (8") (Nº 20-850 µm-Nº 40-425 µm-Nº 50-300 µm), y vaciar el
mortero en el tamiz Nº 20. Repetir la operación con las otras dos porciones
restantes de vidrio, vaciando el mortero cada vez en el tamiz Nº 20. Agitar los
tamices un breve tiempo, luego sacar el vidrio de los tamices Nº 20 y Nº 40, y
moler y tamizar nuevamente como antes. Repetir nuevamente esta operación de
molido y tamizado. Vaciar el recipiente colector, rearmar los tamices, y agitar
en un agitador mecánico durante 5 minutos o a mano durante un tiempo
equivalente. Transferir la porción retenida en el tamiz Nº 50, que debe pesar
no menos de 10 g, a un recipiente cerrado, y almacenar en un desecador hasta
ser usado para el ensayo.
Desparramar la muestra sobre un trozo
de papel parafinado, y pasar un imán sobre la misma para retirar los partículas
de hierro que pudieran pasar a la muestra durante el molido. Transferir la
muestra a un erlenmeyer de vidrio resistente de 250 mL de capacidad, y lavarla
con seis porciones de 30 mL de acetona, agitando cada vez aproximadamente
durante 30 segundos, y decantando cuidadosamente la acetona. Luego del lavado,
la muestra debe quedar libre de aglomeraciones de polvo de vidrio, y la
superficie de los granos debe estar prácticamente libre de partículas finas
adheridas. Secar el erlenmeyer y su contenido durante 20 minutos a 140ºC, transferir los granos a un recipiente para pesar, y enfriar secado.
Procedimiento. Transferir 10.00 g de la muestra preparada, pesada exactamente, a un erlenmeyer de vidrio borosilicato de 250 mL
de capacidad que haya sido previamente tratado con agua para inyectables en un
baño a 90ºC durante no menos de 24 horas o a 121ºC durante 1 hora. Agregar 50.0 mL de agua para inyectables al erlenmeyer y a uno preparado del
mismo modo que servirá de blanco. Tapar los erlenmeyer con tapones de vidrio
borosilicato que hayan sido previamente tratados como los erlenmeyer, y que
calcen bien en la boca de los mismos. Colocar los erlenmeyer en el autoclave, y
cerrarlo perfectamente, dejando la válvula de purga abierta. Calentar hasta que
salga vapor vigorosamente por la válvula, y continuar calentando durante 10
minutos. Cerrar la válvula, y ajustar la temperatura en 121ºC, tardando de 19 a 23 minutos en llegar a la temperatura deseada. Mantener la temperatura a 121 +
2.0ºC. durante 30 minutos, contando el tiempo a partir del momento de alcanzar
esta temperatura. Reducir el calentamiento de modo que el autoclave se enfríe y
llegue a presión atmosférica entre 38 y 46 minutos, venteándose adecuadamente
para prevenir la formación de vacío. Enfriar rápidamente el erlenmeyer con agua
corriente, decantar y transferir el agua a un recipiente convenientemente
limpio, y lavar el vidrio residual molido con cuatro porciones de 15 mL de agua
para inyectables, transfiriendo el agua de lavado a la porción principal.
Agregar 5 gotas de solución de rojo de metilo, y titular inmediatamente con
solución de ácido sulfúrico 0.02 N usando una microbureta. Registrar el volumen
de solución de ácido sulfúrico 0.02 N usada para neutralizar el extracto
obtenido de 10 g de muestra, y corregir el blanco. El volumen utilizado no debe
exceder lo indicado en la Tabla 1.
C-4.3.1.2. Ensayo sobre recipientes de vidrio,
para vidrio Tipo I y II. Lavar cuidadosamente dos veces, 3 o más recipientes de
vidrio seleccionados al azar, con agua para inyectables.
Procedimiento. Llenar cada recipiente hasta el 90%
de su capacidad con agua para inyectables, y tapar los recipientes con papel
aluminio previamente lavado con agua purificada. Colocar los recipientes en el
autoclave y cerrarlo perfectamente, dejando la válvula de purga abierta.
Calentar hasta que salga vigorosamente por la válvula, y continuar calentando
durante 10 minutos. Cerrar la válvula y ajustar la temperatura en 121 ºC, tardando de 19 a 23 minutos en llegar a la temperatura deseada. Mantener la temperatura a 121
+ 2,0ºC durante 60 minutos, contando el tiempo a partir del momento de
alcanzar esa temperatura. Reducir el calentamiento de modo que el autoclave se
enfríe y llegue a la presión atmosférica entre 38 y 46 minutos, venteándose
adecuadamente para prevenir la formación de vacío. Transferir el contenido de
los recipientes en ensayo a un nuevo recipiente con capacidad suficiente.
Tomar una alícuota de 100 mL con una
probeta y transferirla a un erlenmeyer de 250 mL de vidrio borosilicato.
Agregar 5 gotas de solución de rojo de metilo, y titular aún caliente con
solución de ácido sulfúrico 0.02 N. Completar la titulación dentro de los 60
minutos de abierto el autoclave. Registrar el volumen de solución de ácido
sulfúrico 0.02 N usada para neutralizar el extracto de la muestra, y corrigir
un blanco. El volumén no debe exceder lo indicado en la Tabla 1 según el tipo de vidrio en cuestión.
C-4.3.1.4. Tabla I: Tipos de vidrio y límites de
los ensayos
Tipo de vidrio
|
Descripción
|
Tipo de ensayo
|
Límites
Capacidad (mL) Volumen de H2SO4
0.02 N (mL)
|
|
I
|
Vidrio borosilicato neutro
|
C-4. 3. 1. 1
|
Todas
|
1,0
|
|
|
|
C-4. 3. 1. 2
|
Todas
|
0,2
|
|
II
|
vidrio sódico cálcico tratado
|
C-4. 3. 1. 2
|
mayor de 100
|
0,2
|
C-5. CRITERIO PARA LA ACEPTACION O RECHAZO
Los recipientes serán aceptados siempre que cumplan
los requisitos obligatorios (RQ) de esta norma, caso contrario serán rechazados.
ANEXO D
RECIPIENTES PLASTICOS PARA
SOLUCIONES PARENTERALES DE GRAN VOLUMEN
CONTENIDO
D-1
Objetivo
D-2 Definiciones
D-3 Condiciones generales
D-4 Condiciones Específicas
D-5 Métodos de ensayo de los recipientes plásticos
D-6 Criterio para aceptación o rechazo
D-1. OBJETIVO
D-1.1. Esta Norma fija las condiciones exigibles
relativas a los aspectos físicos, químicos y biológicos para los recipientes
plásticos indicados específicamente para el envasado de Soluciones Parenterales
de Gran Volumen (SPGV).
D-1.2. Las exigencias para los recipientes plásticos para
Soluciones Parenterales de Gran Volumen son las siguientes:
a) Asegurar que la calidad de la solución parenteral
se mantenga durante la vida útil del producto;
b) Posibilitar el envasado, la esterilización, el
embalaje, el almacenamiento, el transporte, el manipuleo y la administración de
la solución parenteral en forma segura y eficaz;
c) Evitar la contaminación microbiológica de la
solución;
d) Evitar interacciones físicas, químicas o biológicas,
entre el recipiente y la solución, que afecten la estabilidad de la preparación
y que ocasionen problemas de toxicidad al usuario.
e) Asegurar compatibilidad funcional con los equipos
de infusión.
D-2. DEFINICIONES
A los efectos de esta Norma se adoptan las
definiciones siguientes:
D-2.1. Recipiente plástico. Envase flexible o rígido de
forma y capacidad variada.
D-2.1.1. Recipiente plástico vacío. Recipiente listo, con
inscripción, apropiado para contener SPGV.
D-2.1.2. Recipiente plástico lleno. Recipiente conteniendo
SPGV estéril y apirógena.
D-2.2. Manufactura. Todas las operaciones que intervienen
en la producción del recipiente plástico.
D-2.3. Fabricante. Persona jurídica que realiza las
operaciones de manufactura hasta la obtención del recipiente plástico.
D-2.4. Materias Primas (RQ). Polímeros y aditivos para la
fabricación de recipientes plásticos, que responden a los requisitos de la Farmacopea Europea: "Materiales Plásticos usados para la fabricación de recipientes para
uso parenteral y/o soluciones acuosas para infusiones intravenosas" y
"Recipientes".
En el caso de las materias primas no
incluidas en la Farmacopea Europea, su uso se someterá al acuerdo de los países
integrantes del MERCOSUR.
D-2.5. Agua para inyectables. Agua para fabricación de
SPGV conforme a los requisitos del Anexo B de este reglamento.
D-2.6. Marcado. Inscripción en el recipiente por el
proceso de moldeo o por impresión.
D-2.7. Volumen nominal. Volumen previsto o declarado del
recipiente.
D-3. CONDICIONES GENERALES
D-3.1. Generalidades (RQ). Los recipientes plásticos
deben reunir las siguientes condiciones según especificaciones que se detallan
a continuación:
a) suficientemente transparentes o traslúcidos como
para permitir una inspección visual del contenido contra la luz;
b) fabricados con materiales plásticos exentos de
pigmentos y colorantes.
c) fácilmente vaciables sin insuflarles aire, y
resistentes a la tracción y a la presión.;
d) de paredes uniformes, sin fisuras, rayaduras,
rebabas, burbujas de aire o materiales extraños;
e) compatibles con las SPGV durante el
almacenamiento;
f) apirogénicos, atóxicos y relativamente impermeables
al vapor de agua;
g) que puedan ser cerrados convenientemente, vacíos o
llenos, a fin de evitar la contaminación de los SPGV;
h) provistos de un elemento resistente para la
sustentación y una escala graduada de volumen.
D-3.2. Manufactura. La fabricación, almacenamiento y
transporte de los recipientes plásticos, deben realizarse de acuerdo con las
Buenas Prácticas de Manufactura y las siguientes exigencias:
a) La fabricación de recipientes se debe realizar en
un área limpia.
b) La fabricación por laminación se debe realizar en
un área de clase 10.000, o en un área clase 100.000 con un dispositivo de
eliminación de partículas por electricidad estática, o sobre un flujo laminar
clase 100.
c) El aire utilizado en las máquinas debe ser filtrado
a través de filtros cuya porosidad no sea mayor que 0.45 µm.
D-3.3. Marcado. El marcado de los recipientes debe seguir
las especificaciones del fabricante de las SPGV y la escala graduada debe ser
recalibrada cada vez que se rectifica el molde o se modifican los sistemas de
impresión.
D-3.4. Embalaje. Los recipientes plásticos producidos
deben ser inmediatamente acondicionados en sistemas cerrados, de forma tal que
se cumplan las exigencias de este ReglamentoTécnico.
Los recipientes plásticos cerrados,
fabricados para terceros, deben acondicionarse en bolsas plásticas cerradas, con
un espesor de 0,10 mm como mínimo, con protección externa adicional, por
ejemplo: cajas de cartón o plástico, contenedores flexibles, bolsas de papel o
plástico.
Cada pieza de embalaje debe ser
identificada como mínimo, con la siguiente información:
a) capacidad nominal de los recipientes;
b) número de lote;
c) cantidad;
d) materia prima;
e) fecha de fabricación;
f) número de máquina o molde.
D-4. CONDICIONES ESPECIFICAS
La recepción y el control de las materias primas y
de los recipientes plásticos elaborados por terceros deben ser realizados de
acuerdo con los procedimientos establecidos por las Buenas Prácticas de
Manufactura de SPGV (Anexo A).
D-4.1. Requisitos Físicos
D-4.1.1. Control Visual (RQ). Los recipientes plásticos
deben ser observados en cuanto a su aspecto general y no deben presentar:
a) fallas de soplado (fisuras, rayaduras, rebabas,
escamas, burbujas);
b) inclusiones de materiales, internas y externas;
c) partículas extrañas;
d) sistemas de cierre deficientes;
e) falta de centrado y fallas internas (fisuras,
rayaduras) de las paredes del pico, desde su base hasta el lugar de corte para
su utilización;
f) falta de uniformidad de la unión del molde.
D-4.1.2. Soldadura del pico. El cierre del pico, en las
condiciones del proceso, debe garantizar un perfecto cierre del recipiente.
D-4.1.3. Distribución del material. El recipiente plástico
debe presentar paredes uniformes y con espesores que aseguren la resistencia a
la penetración de microorganismos.
D-4.1.4. Transparencia. El recipiente plástico debe tener
una transparencia tal que posibilite la verificación contra la luz del aspecto
y limpidez de la solución en él contenida, permitiendo la observación de
partículas, turbidez o cambios de color de la solución.
D-4.1.5. Permeabilidad al vapor de agua (RQ)
El recipiente plástico lleno con la
solución parenteral, que puede conservarse también dentro de un envase
protector externo herméticamente cerrado, no debe perder más del 2,5% de la
masa al año a 28 ºC y a 65% de humedad relativa.
D-4.1.6. Resistencia de la base del pico . La base del pico
debe ser resistente a movimientos de flexión sin producirse fisuras, rasgaduras
o pinchaduras, aunque fueran superficiales.
D-4.1.7. Estanqueidad y resistencia a la temperatura y a la
presión interna. El recipiente plástico lleno debe soportar variaciones de
temperatura y de presión sin pérdida de su estanqueidad.
D-4.1.8. Firmeza y estanqueidad de la conexión del pico del
recipiente con el equipo. El pico del recipiente plástico lleno debe permitir
una perfecta conexión con la punta perforante del equipo de infusión, de modo
que no exista vaciamiento y que la punta perforante permanezca asegurada cuando
se la somete a tracción.
D-4.1.9. Resistencia del asa de sustentación. El asa debe permitir
la utilización del recipiente colgado, en las condiciones de uso y durante el
tiempo de infusión de la solución, sin presentar señales de ruptura o
deformación.
D-4.1.10. Resistencia al impacto. Los recipientes llenos
deben resistir el impacto sin presentar rotura, fisura o vaciamiento.
D-4.1.11. Estanqueidad del lugar de inoculación
(RQ). Si se ha previsto un lugar de inoculación en el recipiente plástico, éste
debe permanecer estanco después de la punción y el retiro de la aguja.
D-4.1.12. Adherencia del rótulo (etiqueta)
(RQ). La etiqueta debe adherirse al recipiente de manera que no se separe
durante la vida útil del producto. Deberá cumplir con el ensayo de adherencia.
D-4.1.13. Peso y dimensiones. Los recipientes
plásticos deben poseer un peso y dimensiones de acuerdo con las
especificaciones y límites de tolerancia establecidas por el fabricante.
D-4.2. Requisitos químicos (RQ). Los polímeros y los
recipientes para envases de SPGV deben responder a los requisitos de la Farmacopea Europea: "Materiales Plásticos usados para la fabricación de recipientes para
uso parenteral y/o soluciones acuosas para infusiones intravenosas" y
"Recipientes".
D-4.3. Requisitos biológicos (RQ)
D-4.3.1. Impermeabilidad a los microorganismos (RQ).
Después de su esterilización y durante el almacenamiento, el recipiente
plástico debe garantizar la esterilidad de la solución en el contenido.
D-4.3.2. Toxicidad (RQ). El recipiente plástico no debe
liberar hacia la solución en él contenida, sustancias en cantidades capaces de
ejercer efectos tóxicos.
Los componentes de adhesivos, de colas
de los rótulos y de tintas de impresión, no deben atravesar las paredes del
recipiente.
D-4.3.3. Sustancias piretógenas (RQ). El recipiente
plástico no debe liberar hacia la solución en él contenida sustancias capaces
de ejercer efectos piretogénicos.
D-5. METODOS DE ENSAYO DE LOS RECIPIENTES PLASTICOS
Los ensayos siguientes deberán realizarse sobre
muestras obtenidas siguiendo un plan aceptable de muestreo. En cada caso deberá
establecerse el criterio de aceptabilidad y la tolerancia.
D-5.1. Ensayos físicos
D-5.1.1. Control visual (RQ). Los envases examinados no
deberán presentar a simple vista los efectos descriptos en D-4.1.1.
D-5.1.2. Soldadura previa del pico. Los picos de los
recipientes plásticos llenos, deben ser cerrados simulando el procedimiento
industrial, observando si existe una soldadura perfecta con cierre hermético
del pico.
D-5.1.3. Distribución del material. El espesor de las
paredes debe medirse en las partes superior, media e inferior del recipiente
plástico.
D-5.1.4. Transparencia. Llenar un envase con un volumen
igual a su capacidad nominal con solución opalescente primaria diluida 1 en 200
en el caso de envases de polietileno o polipropileno, 1 en 400 para otros
envases. La turbidez de la suspensión debe ser perceptible cuando se observa a
través del envase y se lo compara con un envase similar lleno con agua.
Reactivos
Solución de sulfato de hidrazina
Disolver 1.0 g de sulfato de hidrazina en agua y diluir a 100 mL con el mismo solvente.
Dejar en reposo durante 4 a 6 horas.
Solución de hexametilentetramina
Disolver 2,5 g de hexametilentetramina en 25 mL de agua en un matraz de vidrio con tapón, de 100 mL de
capacidad.
Solución opalescente primaria
Agregar a la solución de
hexametilentetramina contenida en un matraz, 25,0 mL de solución de sulfato de
hidrazina. Mezclar y dejar en reposo por 24 horas. Esta solución es estable
durante 2 meses, cuando se la almacena en recipientes de vidrio libres de defectos
superficiales.
La suspensión no debe adherirse al
vidrio y debe ser bien mezclada antes de ser usada.
D-5.1.5. Permeabilidad al vapor de agua (RQ). Los
recipientes plásticos llenos con la solución parenteral, que pueden conservarse
también dentro de un envase protector externo herméticamente cerrado, deberán
almacenarse a 28ºC y 65% de humedad relativa durante 3 meses. Cada 7 días a
partir del comienzo del ensayo, deberán pesarse a fin de establecer la curva de
la eventual pérdida de peso por permeabilidad al vapor de agua. Al finalizar el
ensayo la pérdida de peso no debe exceder el 0,625% (2,5% al año) en las
condiciones especificadas por el fabricante para el espesor de la pared del
recipiente plástico que contiene la solución y el espesor de la pared del
envase protector externo.
D-5.1.6. Resistencia de la base del pico. La parte superior
de los recipientes plásticos (pico) debe doblarse 10 veces hacia la izquierda y
hacia la derecha, formando un ángulo de 30 grados respecto de su posición inicial.
En la base de los picos no deben formarse fisuras, pinchaduras o rasgaduras,
aunque fueran superficiales.
D-5.1.7. Estanqueidad y resistencia a la temperatura y a la
presión interna. Colocar los recipientes llenos durante 24 horas a una
temperatura entre -5ºC y +5ºC, y a continuación entre 50 y 55ºC. Después de llevarlos a temperatura ambiente, colocar los recipientes entre dos placas paralelas
y someterlos a una presión interna de 100 KPa durante 10 minutos a 20ºC. No deben producirse pérdidas de líquido.
D-5.1.8. Firmeza y estanqueidad de la conexión del pico del
recipiente con el equipo. Conectar las puntas perforantes de los equipos a los
picos de los recipientes plásticos llenos simulando las condiciones de uso.
Colgar los conjuntos del soporte de
infusión, y aplicar a las cámaras de goteo una fuerza de tracción dirigida
hacia abajo de 10 N durante 5 horas. Los equipos de infusión no deben
desprenderse y la estanqueidad debe garantizarse.
D-5.1.9. Resistencia del asa de sustentación. A los recipientes
llenos, colgados, aplicar una fuerza longitudinal mínima de 25 N durante 5
horas. Las asas de sustentación no deben presentar señales de rotura o de
deformación.
D-5.1.10. Resistencia al impacto. Dejar caer
los recipientes plásticos llenos desde una altura de 2 m sobre una superficie lisa y rígida. Este impacto no debe causar estallido, ruptura, fisura o
vaciado en cualquier lugar de los recipientes.
D-5.1.11. Estanqueidad del lugar de inoculación
(RQ). Punzar los lugares de inoculación de los recipientes vacíos y cerrados,
con una aguja de 0,6 mm de diámetro externo.
Retirar la aguja y verificar la
estanqueidad de los puntos de inoculación, sumergiendo los recipientes en agua
y sometiéndolos a una presión interna de 20 KPa durante 15 segundos.
No debe haber pérdidas de aire.
D-5.1.12. Adherencia del rótulo (RQ). Mantener
no menos de 5 envases a temperatura ambiente durante 5 días. Luego sumergirlos
en agua a 24º C + 2º C durante 48 horas. Al final del ensayo todos los rótulos
deben permanecer adheridos a los recipientes.
D-5. 1.13. Peso y dimensiones. El peso y
dimensiones de los envases deben estar dentro de los límites de tolerancia
establecidos en D.4.1.13.
D-5.2. Ensayos químicos (RQ). Para los ensayos químicos de
los polímeros y de los recipientes plásticos se debe adoptar la metodología
descripta en la Farmacopea Europea: "Materiales plásticos usados para la
fabricación de recipientes para uso parenteral y/o soluciones acuosas para
infusiones intravenosas" y "Recipientes".
D-5.3. Ensayos biológicos (RQ)
D-5.3.1. Impermeabilidad a los microorganismos (RQ). Llenar
4 recipientes plásticos hasta su volumen nominal con medio de cultivo caldo
triptona-soja, y esterilizar; o usar un proceso de llenado estéril.
Incubar los recipientes durante 48 horas
a 37º C, de modo que si hay contaminación, ésta pueda visualizarse. Colocar los
recipientes en frascos de vidrio con tapa y conteniendo el mismo medio de
cultivo usado anteriormente, de modo que partes de los recipientes plásticos
queden inmersos.
Inocular el medio de cultivo del
frasco de vidrio con un cultivo de serratia marcescens en caldo e incubar a
30-32º C durante 10 días.
Preparar como control positivo un
recipiente plástico como el indicado más arriba, inoculado con 1 mL del cultivo
bacteriano usado en el ensayo. El recipiente control no se coloca en el frasco
de vidrio como los otros cuatro recipientes, pero se incuba durante 10 días a
30 - 32º C.
El medio de cultivo contenido en el
recipiente control debe presentar nítida turbidez, mientras que los medios de
cultivo contenidos en los recipientes plásticos en ensayo deben permanecer
límpidos.
D-5.3.2. Ensayos de toxicidad (RQ). Utilizar la metodología
descripta en el anexo L Ensayos biológicos
L.5. Ensayos de reactividad biológica
L.5.1. Ensayo de reactividad biológica "in
vitro"
L.5.2. Ensayo de reactividad biológica "in
vivo"
D-5.3.3. Sustancias piretógenas (RQ)
D-5.3.3.1. Obtención
del extracto
Tomar una muestra de 25 cm x 25 cm (625 cm2) del recipiente plástico, verificando que no posea etiquetas ni
impresiones. El tamaño de la muestra equivale a 1250 cm2 de
superficie total teniendo en cuenta ambas caras.
Cuando las dimensiones del recipiente
no permitan tomar en un solo trozo el material en las medidas indicadas, tomar
tantos trozos como sea necesario para obtener una superficie total de 1250 cm2.
Cortar la muestra en trozos de 2 cm x 5 cm (10 cm2). Lavar dos veces con agua destilada.
Introducir en un matraz de
borosilicato que contenga 250 mL de solución isotónica de cloruro de sodio.
Colocar en autoclave a 121° C durante
60 minutos.
Enfriar y llevar el volumen a 250 mL
con agua estéril y apirogénica.
Efectuar paralelamente un ensayo en
blanco con la misma cantidad de solución isotónica de cloruro de sodio.
Notas:
1) No tiene importancia que durante el proceso de
autoclavado los trozos de material plástico se adhieran ligeramente entre sí.
2) Cuando el material plástico sea sensible al calor,
calentar el matraz con su contenido a 70º C durante 24 horas o 50º C durante 72
horas.
3) La extracción puede realizarse con un recipiente
plástico completo manteniendo la relación: 250 mL de agua destilada por cada
1250 cm2 de superficie total del material.
4) Cuando sea necesario un mayor volumen de extracto
que el indicado en el procedimiento de extracción (250 mL) para poder
cumplimentar los distintos ensayos, tomar mayor cantidad de muestra respetando
la relación indicada en 3).
D-5.3.3.2. Ensayos. Utilizar la Metodología descripta en el anexo L Ensayos Biológicos
L.2. Ensayo de piretógenos
L.3. Ensayo de endotoxinas bacterianas.
Límite de endotoxinas: 0,5 E U/mL
D-6. CRITERIO PARA ACEPTACION O RECHAZO
Los recipientes serán aceptados siempre que cumplan
los requisitos obligatorios (RQ) de esta norma, en caso contrario serán
rechazados.
ANEXO E
ESPECIFICACIONES Y CONTROL DEL
PRODUCTO TERMINADO
CONTENIDO
E-1
OBJETIVO
E-2
PRODUCTOS TERMINADOS
E-3
REQUERIMIENTOS GENERALES
E-4
REACTIVOS
E-1.
OBJETIVO
Esta norma establece especificaciones para los
productos terminados y los procedimientos respectivos para su control.
E-2.
PRODUCTOS TERMINADOS
E-2.1. SOLUCIÓN DE CLORURO DE SODIO INYECTABLE.
La solución de Cloruro de Sodio
Inyectable es una solución estéril de Cloruro de Sodio en Agua para
Inyectables. No contiene agentes antimicrobianos. Contiene no menos del 95.0% y
no más del 105.0% de la cantidad indicada de ClNa en el rótulo.
E-2.1.1. Especificaciones y Procedimientos de Control.
E-2.1.1.1. Envasado y Conservación:
Conservar en envases monodosis de plástico o vidrio, éste último
preferentemente de tipo I o tipo II.
E-2.1.1.2. Rotulado: El rótulo responderá
a lo especificado en el Reglamento Técnico para SPGV punto 8.
E-2.1.1.3. Identificación: Responde a los
ensayos para Sodio (USP XXII-191) y para Cloruro (USP XXII-191).
E-2.1.1.4. Sustancias piretógenas: Podrá
optarse por uno de los siguientes ensayos.
a) Ensayo de piretógenos: Cumple con los
requerimientos del ensayo de Piretógenos (Anexo L.2). (NOTA: Diluir con Agua para
Inyectables aquellas soluciones que contengan más del 0.9% de cloruro de sodio
para dar una concentración del 0.9%).
b) Ensayo de endotoxinas bacterianas: Se
llevará a cabo de acuerdo a lo descripto en "Ensayo de endotoxinas
bacterianas" Anexo L.3. No deberá contener más de 0.5 UE/mL cuando la
concentración de cloruro de sodio se encuentre entre 0,5% y 0,9%. Para
soluciones con concentraciones de cloruro de sodio entre 3% y 24,3%, éstas no
podrán contener más de 3,6 UE/mL.
E-2.1.1.5. pH (USP) entre 4,5 y 7,0.
E-2.1.1.6. Partículas extrañas: Cumple
con el ensayo de "Partículas extrañas" (E. 3. 1. 10).
E-2.1.1.7. Hierro (USP XXII-24 1): Diluir
5,0 mL de solución con agua hasta 45 mL, y agregar 2 mL de ácido clorhídrico:
el límite es 2 ppm.
E-2.1.1.8. Metales Pesados (USP XXII-23 1
Método 1): Colocar un volumen de solución equivalente a 1.0 g de cloruro de sodio, en un vaso adecuado, si es necesario evaporar a un volumen de
aproximadamente 20 mL, agregar 2 mL de ácido acético IN, luego diluir con agua
a 25 mL. Proceder como está indicado, excepto que se debe usar 1 mL de Solución
Standard de Plomo (10 ug de Pb) en la Preparación Standard y en la Preparación Control: el límite es de 0.001%, en base a la
cantidad de cloruro de sodio.
E-2.1.1.9. Otros requerimientos: Cumple
con los "Requerimientos generales para las SPGV" (E.3. 1)
E-2.1.1.10. Valorización: Pipetear un
volumen de Solución de Cloruro de Sodio para Inyectables, equivalente a
aproximadamente 90 mg de cloruro de sodio, en una cápsula de porcelana, y
agregar 140 mL de agua y 1 mL de diclorofluoresceína SR. Mezclar, y titular con
nitrato de plata 0.1N SV hasta que flocule el cloruro de plata y la mezcla
adquiera un color rosa débil. Cada mL de nitrato de plata 0.1N es equivalente a
5.844 mg de NaCI.
E-2.2. SOLUCIÓN DE DEXTROSA INYECTABLE:
La Solución de Dextrosa Inyectable es una solución estéril de
dextrosa en Agua para inyectables. Contiene no menos del 95.0% y no más del
105.0 % de la cantidad indicada en el rótulo de C6H12O6H2O.
No contiene agentes antimicrobianos.
E-2.2.1. Especificaciones y Procedimientos de Control
E-2.2.1.1. Envasado y conservación:
Conservar en envases monodosis de plástico o vidrio, éste último
preferentemente de tipo I o tipo II.
E-2.2.1.2. Rotulado: El rótulo responderá
a lo especificado en el Reglamento Técnico para SPGV punto 8.
E-2.2.1.3. Identificación: Responde a la
prueba de Identificación detallada para Dextrosas en Anexo B-3.1.
E-2.2.1.4. Sustancias piretógenas: Podrá
optarse por uno de los siguientes ensayos:
a) Ensayo de piretógenos: Cumple con los
requerimientos del ensayo de piretógenos (Anexo L.2). (NOTA. Diluir con Agua
para Inyectables las soluciones que contengan más del 10% de dextrosa para
llevar a una concentración del 10%).
c) Ensayo de endotoxinas bacterianas: Se
llevará a cabo de acuerdo a lo descripto en "Ensayo de endotoxinas
bacterianas" Anexo L.3. No deberá contener más de 0.5 UE/mL cuando la
concentración de dextrosa sea menor al 5%. Para soluciones con concentraciones
de dextrosa entre el 5 % y el 70%, éstas no podrán contener más de 10,0
UE/gramo de dextrosa. NOTA: Previo al análisis, diluir las soluciones que
contengan más del 10% de dextrosa para llevar a una concentración del 10%.
E-2.2.1.5. pH (USP XXII-791). Entre 3.5 y
6.5 determinado en una alícuota a la cual se han agregado 0.30 mL de solución
saturada de cloruro de potasio cada 100 mL y que previamente se ha diluido con
agua, si es necesario, a una concentración de no más del 5% de dextrosa.
E-2.2.1.6. Partículas extrañas. Cumple el
ensayo de "Partículas extrañas" (E-3. 1. 10).
E-2.2.1.7. Metales Pesados (USP XXII-23
1). Transferir un volumen de solución, equivalente a 4.0 g de dextrosa, a un recipiente adecuado, y ajustar el volumen a 25 mL por evaporación o agregado
de agua, según sea necesario: el límite es de 0.0005 C%, donde C es la cantidad
rotulada, en gramos de, C6H12O6H2O
por mL de Solución.
E-2.2.1.8. Hidroximetilfurfural y
sustancias relacionadas:
Diluir un volumen exactamente medido de solución, equivalente a 1.0 g de C6H12O6H2O, con agua hasta 250.0 mL. Determinar
la absorbancia de esta solución en una celda de 1 cm a 284 mm, con un espectrofotómetro adecuado, usando agua como blanco: la absorbancia no debe ser
mayor de 0.25.
E-2.2.1.9. Otros requerimientos: Cumple con los "Requerimientos
generales para SPGV" (E. 3. 1).
E-2.2.1.10. Valoración: Transferir un volumen exactamente
medido de Solución de Dextrosa Inyectable, que contenga 2 a 5 g de dextrosa, a un matraz aforado de 100 mL. Agregar 0,2 mL de hidróxido de amonio 6 N, llevar
a volumen con agua y mezclar. Determinar la rotación angular en un tubo
polarimétrico adecuado a 25º (Ver Rotación Optica (USP XXII-781)). La rotación
observada, en grados, multiplicada por 1,0425 A, en la cual A es el cociente entre 200 y la longitud en mm, del tubo de polarímetro empleado, representa el peso,
en g, de C6H12O6H2O en el volumen
de solución tomado.
E-2.3. SOLUCIÓN INYECTABLE DE DEXTROSA Y CLORURO DE
SODIO.
La Solución Inyectable de Dextrosa y Cloruro de Sodio es una solución
estéril de Dextrosa y Cloruro de Sodio en Agua para Inyectables. Contiene no
menos del 95.0% y no más del 105.0% de la cantidad indicada en el rótulo de C6H12O6H2O
y de NaCl. No contiene agentes antimicrobianos.
E-2.3.1. Especificaciones y Procedimientos de Control
E-2.3.1.1. Envasado y Conservación. Conservar
en envases monodosis de plástico o vidrio, éste último preferentemente de tipo
I o tipo II.
E-2.3.1.2. Rotulado. El rótulo responderá
a lo especificado en el Reglamento Técnico para SPGV punto 8.
E-2.3.1.3. Identificación. Responde al
ensayo de Identificación indicado en Dextrosa ANEXO B-3. 1, y a los ensayos
para Sodio (USP XXII-191) y para Cloruro (USP XXII-191).
E-2.3.1.4. Sustancias piretógenas. Podrá
optarse por uno de los siguientes ensayos:
a) Ensayo de piretógenos: Cumple con los
requerimientos del ensayo de piretógenos (Anexo L.2), establecidos en Solución
de Dextrosa Inyectable.
b) Ensayo de endotoxinas bacterianas: Se llevará a
cabo de acuerdo a lo descripto en "Ensayo de endotoxinas bacterianas"
Anexo L.3. No deberá contener más de 10,0 UE/gramo de dextrosa.
E-2.3.1.5. pH (USP XXII-791). Entre 3.5 y 6.5,
determinado en una alicuota con agua, si es necesario, hasta una concentración
de no más del 5% de dextrosa.
E-2.3.1.6. Hidroximetilfurfural y
sustancias relacionadas.
Diluir un volumen exactamente medido de solución, equivalente a 1.0 g e C6H12O6H2O con agua hasta 500.000.0 mL.
Determinar la absorbancia de esta solución en una celda de 1 cm a 284 nm, con un espectrofotómetro adecuado, usando agua como blanco: la absorbancia es no más
de 0.25.
E-2.3.1.7. Otros requerimientos. Cumple
con los "Requerimientos generales para las SPGV" (E.3. 1).
E-2.3.1.8. Valoración de dextrosa.
Transferir un volumen exactamente medido de Solución Inyectable de Dextrosa y
Cloruro de Sodio, que contiene de 2 a 5 g de dextrosa, a un frasco volumétrico de 100 mL. Agregar 0.2 mL de hidróxido de amonio 6 N, llevar a volumen con
agua, y mezclar. Determinar la rotación angular en un tubo polarimétrico
adecuado a 25º (Ver Rotación Optica (USP XXII-781)). La rotación observada, en
grados, multiplicada por 1.0425 A, donde A es la relación 200 dividido la
longitud, en mm, del tubo polarimétrico empleado, representa el peso, en g, de
C6H12O6H2O en el volumen de
solución tomado.
E-2.3.1.9. Valoración de Cloruro de Sodio.
Transferir un volumen exactamente medido de Solución Inyectable de Dextrosa y
Cloruro de Sodio, equivalente a aproximadamente 90 mg de cloruro de sodio, a
una cápsula de porcelana, y agregar 140 mL de agua y 1 mL de
diclorofluoresceína SR. Mezclar y titular con nitrato de plata 0.1N SV hasta
que flocule el cloruro de plata y la mezcla adquiera un débil color rosa. Cada
mL de nitrato de plata 0.1N es equivalente a 5.844 mg de NaCl.
E-2.4.
AGUA ESTERIL PARA INYECTABLES
El Agua Estéril para Inyectables es
Agua para Inyectables esterilizada y adecuadamente envasada. No contiene
agentes antimicrobianos u otra sustancia agregada.
E-2.4.1. Especificaciones y Procedimientos de
Control
E-2.4.1.1. Envasado y Conservación.
Conservar en envases monodosis de plástico o vidrio, éste último
preferentemente de tipo I o tipo II.
E-2.4.1.2. Rotulado. El rótulo responderá
a lo especificado en el Reglamento Técnico para SPGV punto 8.
E-2.4.1.3. Sustancias piretógenas. Podrá
optarse por uno de los siguientes ensayos.
a) Ensayo de piretógenos: Cumple con los
requerimientos del ensayo de piretógenos (Anexo L. 2). NOTA: Isotonizar el agua
antes de su inyección.
b) Ensayo de endotoxinas bacterianas: Se llevará a
cabo de acuerdo a lo descripto en "Ensayo de endotoxinas bacterianas"
Anexo L. 3. No deberá contener más de 0,25 UE/mL.
E-2.4.1.4. Esterilidad. Cumple con los
requerimientos de Ensayos de Esterilidad (Anexo L.4).
E-2.4.1.5 Partículas extrañas. Cumple con el ensayo de
"Partículas extrañas" (E. 3. 1. 10).
E-2.4.1.6. Amoníaco. Usar 100 mL de Agua
Estéril para Inyectables como solución de ensayo.
A. 100 mL de la solución de ensayo,
agregar 2 mL de ioduro mercúrico potásico alcalino SR: cualquier color amarillo
producido inmediatamente no debe ser más oscuro que el de un control que
contiene 30 ug de NH3 agregado a Agua para inyectables (conductividad no mayor
de 0,.15 umho/cm a 251ºC). Límite 0,3 ppm.
E-2.4.1.7. Cloruro. A 20 mL en un tubo
para comparación de color agregar 5 gotas de ácido nítrico y 1 mL de nitrato de
plata SR, y mezclar suavemente: cualquier turbidez formada dentro de los 10
minutos no debe ser mayor que la producida en un control tratado en forma
similar realizado con 20 mL de Agua químicamente pura (C.4.3.1.1) que contenga
10 ug de CI (0.5 ppm), observando las soluciones hacia abajo sobre una
superficie oscura con luz lateral.
E-2.4.1.8. Sustancias Oxidables. A 100 mL
agregar 10 mL de ácido sulfúrico 2 N, y calentar a ebullición. Para Agua
Estéril para Inyectables en recipientes de vidrio de hasta 50 mL, agregar 0.4
mL de permanganato de potasio 0.1 N, y hervir durante 5 minutos, para volúmenes
más grandes, agregar 0.2 mL de permanganato de potasio 0.12 N y hervir 5
minutos: el color rosa no desaparece completamente.
E-2.4.1.9. Residuo por evaporación.
Proceder como se indica en el ensayo de "Residuos por evaporación"
para Agua para inyectables (B-2. 1. 8), el límite para Agua Estéril para
Inyectables es 0.002%.
E-2.4.1.10 Otros requerimientos. Cumple
con los requerimientos de los ensayos de pH (USP XXII-791): entre 5.0 y 7.0,
determinado potenciométricamente en una solución preparada por el agregado de
0.30 mL de solución saturada de cloruro de potasio a 100 mL de la muestra a
analizar.
Sulfato: a 100 mL agregar 1 mL de
cloruro de bario SR: no se produce turbidez.
Calcio: a 100 mL, agregar 2 mL de
oxalato y amonio SR: no se produce turbidez.
Dióxido de Carbono: a 25 mL agregar 25
mL de hidróxido de calcio TS: la mezcla permanece clara.
Metales Pesados: ajustar 40 mL de Agua
Estéril para Inyectables con ácido acético 1N a un pH de 3.0 a 4.0 (usando un papel indicador de pH de poco rango), agregar 10 ml de sulfuro de hidrógeno SR
recientemente preparado, y dejar descansar el líquido 10 minutos: el color del
líquido, cuando se observa hacia abajo sobre una superficie blanca, no debe ser
más oscuro que el color de una mezcla de 50 mL de la misma Agua Estéril para
Inyectables con la misma cantidad de ácido acético 1N como se agregó a la
muestra, usándose sendos tubos para comparación de color para el ensayo.
E-2.5. SOLUCIÓN DE MANITOL INYECTABLE.
La Solución de Manitol Inyectable es una solución estéril, que
puede ser sobresaturada, de Manitol en Agua para Inyectables. Puede requerir
calentamiento o autoclavado antes de usar si ha ocurrido cristalización.
Contiene no menos del 95.0% y no más de 105.0% de la cantidad rotulada de
C6H14O6. No contiene agentes antimicrobianos.
E-2.5.1. Especificaciones y Procedimientos de
Control
E-2.5.1.1. Envasado y Conservación.
Conservar en envases monodosis de plástico o vidrio, éste último
preferentemente de tipo I o tipo II.
E-2.5.1.2. Rotulado. El rótulo
responderá a lo especificado en el Reglamento Técnico par SPGV punto 8.
E-2.5.1.3. Identificación. Evaporar a
sequedad en un baño de vapor una alícuota de la Solución Inyectable, y secar el residuo a 105º durante 4 horas: el residuo responde a los
ensayos de Identificación que figuran en Manitol.
E-2.5.1.4. Rotación Específica (USP
XXII-781). Transferir a un frasco volumétrico un volumen exactamente medido de la Solución Inyectable, equivalente a aproximadamente 1 g de manitol determinado por la "Valoración", cumple con los requerimientos de la prueba para Rotación
Específica para Manitol (B-3. 15. 1. 5).
E-2.5.1.5. Sustancias piretógenas.
Cumple con los requerimientos del ensayo de Piretógenos (L.2). NOTA: diluir, si
es necesario, con Agua para Inyectables para contener no más del 10% de
C6H14O6.
E-2.5.1.6. pH (USP XXII-791). Entre 4.5
y 7.0, determinado potenciométricamente, en una solución preparada por agregado
de 0.30 mL de solución saturada de cloruro de potasio a 100 mL de Solución
Inyectable de Manitol, previamente diluida a una concentración no mayor de 5%
si es necesario.
E-2.5.1.7. Partículas extrañas. Cumple con el
ensayo de "Partículas extrañas" (E. 3. 1. 10).
E-2.5.1.8. Otros requerimientos.
Cumple con los "Requerimientos generales para las SPGV" (E.3.1).
E-2.5.1.9. Valoración. Transferir un
volumen exactamente medio de la Solución Inyectable de Manitol, equivalente a aproximadamente 1 g de manitol, a un matraz aforado de 1000 mL, agregar agua hasta
volumen y mezclar. Transferir 4.0 mL de esta solución a un frasco cónico de 250
mL, y agregar 50.0 mL de un reactivo preparado mezclando 40 mL de ácido
sulfúrico 2 N con 60 mL de una solución de periodato de potasio (1 en 1000)
acidificada con 3 a 5 gotas de ácido sulfúrico. Calentar la solución en baño de
vapor durante 15 minutos, enfriar a temperatura ambiente y agregar 1 g de ioduro de potasio. Dejar descansar 5 minutos, y titular con tiosulfato de sodio 0.02 N SV,
agregando 3 mL de almidón SR cuando se acerque el punto final. Realizar la
determinación en un blanco, usando agua en lugar de la Solución de Manitol Inyectable, y considerar la diferencia en los volúmenes consumidos. Cada
mL de la diferencia en volumen del tiosulfato de sodio 0,02 N consumido es
equivalente a 0.3643 mg de C6H14O6.
E-2.6. SOLUCIÓN DE LACTATO DE SODIO
INYECTABLE.
La Solución de Lactato de Sodio Inyectable es una Solución
estéril de Lactato de Sodio en Agua para Inyectables, o una Solución estéril de
Acido Láctico en Agua para Inyectables preparada con la ayuda de Hidróxido de
Sodio. Contiene no menos del 95.0% y no más del 110.0 % de la cantidad indicada
en el rótulo de C3H5NaO3.
E-2.6.1. Especificaciones y Procedimientos de Control.
E-2.6.1.1. Envasado y Conservación:
Conservar en envases monodosis de plástico o vidrio, éste último
preferentemente de tipo I o tipo II.
E-2.6.1.2. Rotulado. El rótulo responderá
a lo especificado en el Reglamento Técnico para SPG punto 8.
E-2.6.1.3. Identificación:
A) Extender 2 mL de la Solución Inyectable sobre 5 mL de una solución 1 en 100 de catecol en ácido sulfúrico: se
produce un color rojo profundo en la zona de contacto.
B) A 2 mL de la Solución Inyectable agregar 5 mL de ácido sulfúrico 2 N y 2 mL de permanganato de potasio SR
y calentar: se desarrolla olor a acetaldehído.
E-2.6.1.4. Sustancias piretógenas. Podrá optarse por uno de los siguientes
ensayos:
a) Ensayo de piretógenos: Cumple con los
requerimientos del ensayo de piretógenos (Anexo L. 2). (NOTA:
Diluida, si es necesario, con agua para inyectables a aproximadamente 0.16 M (20 mg por ml)).
b) Ensayo de endotoxinas bacterianas: Se llevará a
cabo de acuerdo a lo descripto en "Ensayo de endotoxinas bacterianas"
Anexo
L.3. No deberá contener más de 2,0
UE/mEq.
E-2.6.1.5. pH (USP XXII-791). Entre 6. 0
y 7. 3, diluyendo una alicuota de la solución Inyectable con agua, si es
necesario, a aproximadamente 0.16 M (20 mg por mL).
E-2.6.1.6. Partículas extrañas. Cumple
con el ensayo de "Partículas extrañas" (E.3.1.10).
E-2.6.1.7. Metales Pesados (USP XXII-23). Evaporar un volumen de la Solución Inyectable, equivalente a 2.0 g de lactato de sodio, a 5 mL, y diluir con ácido
acético 1 N a 25 mL: el límite es de 0,001%.
E-2.6.1.8. Otros Requerimientos. Cumple
con los "Requerimientos generales para las SPGV" (E. 3. 1).
E-2.6.1.9. Valoración. Pipetear dentro de
un recipiente pequeño un volumen de Solución de Lactato de Sodio Inyectable,
equivalente a aproximadamente 300 mg de lactato de sodio, y evaporar a
sequedad. Agregar al residuo 60 mL de una mezcla 1 en 5 de anhídrido acético en
ácido acético glacial, y agitar hasta que el residuo esté completamente
disuelto. Titular con ácido perclórico 0.1N S.V. determinando el punto final
potencialmente. Realizar la determinación en un blanco y hacer cualquier
corrección que fuera necesaria. Cada mL de ácido perclórico 0.1N es equivalente a 11.21 mg de C3H5NaO3.
E-2.7. SOLUCIÓN DE RINGER INYECTABLE.
La Solución de Ringer Inyectable es una solución estéril de
Cloruro de Sodio, Cloruro de Potasio y Cloruro de Calcio en Agua para Inyectables.
Contiene, no menos del 95% y no más del 105% de la cantidad rotulada de ClNa,
no menos del 90% y no más del 110% de la cantidad rotulada de ClK y no menos
del 90% y no más del 110% de la cantidad rotulada de Cl2Ca.2H2O.
La Solución de Ringer Inyectable no contiene agentes antimicrobianos. Disolver
las tres sales en el Agua para Inyectable, filtrar hasta que la solución esté
clara, colocar en recipientes adecuados, y esterilizar.
E-2.7.1. Especificaciones y Procedimientos de Control
E-2.7.1.1. Envasado y Conservación:
Conservar en envases monodosis de plástico o vidrio, éste último
preferentemente de tipo I o tipo II.
E-2.7.1.2. Rotulado. El rótulo responderá
a lo especificado en el Reglamento Técnico para SPGV punto 8.
E-2.7.1.3. Identificación. Responde a los
ensayos para Sodio (USP XXII-191) y para Cloruro (USP XXII-191), y cuando se
concentra a la mitad de su volumen original, al ensayo de Calcio (USP XXII-191)
y al ensayo de la llama para Potasio (USP XXII-191).
E-2.7.1.4. Sustancias piretógenas. Podrá
optarse por uno de los siguientes ensayos:
a) Ensayo de piretógenos: Cumple con los
requerimientos del ensayo de piretógenos (Anexo L.3).
b) Ensayo de endotoxinas bacterianas: Se llevará a
cabo de acuerdo a lo descrito en "Ensayo de endotoxinas bacterianas"
Anexo L.3. No deberá contener más de 0,5 UE/mL.
E-2.7.1.5. pH (USP XXII-791). Entre 5. 0 y 7. 5.
E-2.7.1.6. Metales Pesados (USP
XXII-231). Evaporar 67 mL a un volumen de alrededor de 20 mL, agregar 2 mL de
ácido acético 1 N, y diluir con agua a 25 mL: el límite es de 0.3 ppm.
E-2.7.1.7. Otros requerimientos. Cumple
con los "Requerimientos generales para las SPGV" (E.3.1.).
E-2.7.1.8. Valorización de Calcio.
Pipetear 50 mL de la Solución de Ringer Inyectable en un vaso de 250 mL,
agregar 15 mL de hidróxido de sodio 1N y 300 mg de azul de hidroxi naftol
triturado, y titular inmediatamente con etilenediaminotetraacetato disódico 0.005 M SV a un punto final azul profundo. Cada mL de etilendiaminotetraacetato disódico 0.005 M es equivalente a 200.4 Og de Ca++.
E-2.7.1.9. Valoración de Potasio
Solución. Stock Standard
Disolver 190.7 mg de cloruro de
potasio, previamente secado a 105º durante 2 horas, en 50 mL de agua,
transferir a un frasco volumétrico de 1000 mL, diluir con agua hasta volumen, y
mezclar. Cada mL de esta solución contiene 100 Og de potasio.
Preparaciones standard.
Disolver 1.093 g de cloruro de sodio en 100.0 mL de agua, y transferir 10 mL de esta solución a cada uno de
cinco frascos de 100 mL que contienen 10.0 mL de una solución de un agente
humectante no iónico adecuado (1 en 500). Diluir el contenido de uno de los
frascos con agua hasta volumen para obtener un blanco. A los restantes frascos,
agregar, respectivamente, 5.0, 10.0, 15.0 y 20.0 mL de la Solución Stock Standard, diluir con agua hasta volumen, y mezclar.
Preparación de ensayo.
Pipetear 10 mL de la Solución de Ringer Inyectable en un frasco volumétrico de 100 mL, agregar 10.0 mL de una
solución de un agente humectante adecuado (1 en 500), diluir con agua hasta
volumen, y mezclar.
Gráfico
standard
Colocar un fotómetro de llama adecuado
en transmitancia máxima a una longitud de onda de aproximadamente 766 mm. Ajustar el instrumento a 0% de transmitancia con el blanco y a 100% de transmitancia con la
más concentrada de las preparaciones standard. Leer el porcentaje de
transmitancia de las otras Preparaciones Standard, y graficar las
transmitancias versus la concentración de potasio.
Procedimiento.
Ajustar el instrumento como se indica
en Gráfico Standard, leer el porcentaje de transmitancia de la Preparación de Prueba, y calcular el contenido de potasio, en mg por 100 mL de la Solución de Ringer Inyectable.
E-2.7.1.10. Valoración de Sodio.
Solución Stock standard.
Disolver 254.2 mg de cloruro de sodio,
previamente secado a 105º durante 2 horas, en 50 mL de agua, transferir a un
frasco volumétrico de 1000 mL, diluir con agua hasta volumen, y mezclar. Cada
mL de esta solución contiene 100 Og de sodio.
Preparaciones standard.
Transferir a cada uno de cinco frascos
volumétricos de 100 mL de una solución de un agente humectante no iónico
adecuado (1 en 500). Diluir el contenido de uno de los frascos con agua hasta
volumen para obtener un blanco. A los restantes frascos agregar, respectivamente,
5.0, 10.0, 15.0 y 20.0 mL de la Solución Stock Standard, diluir con agua hasta volumen, y mezclar.
Preparación de Ensayo.
Pipetear 5 mL de Solución de Ringer
Inyectable en un frasco volumétrico de 1000 mL que contiene 100 mL de una
solución de un agente humectante adecuado (1 en 500), diluir con agua hasta
volumen y mezclar.
Procedimiento.
Proceder como se indica en Gráfico
Standard y en Procedimiento en el Ensayo para Potasio, colocando el fotómetro
de llama en transmitancia máxima a una longitud de onda de aproximadamente 589 mm, en lugar de aproximadamente 766 mm. Calcular el contenido de sodio, en mg por 100 mL, de la Solución de Ringer Inyectable.
E-2.7.1.11. Valoración de Cloruro.
Pipetear 10 mL de la Solución de Ringer Inyectable en una cápsula de porcelana, y agregar 140 mL de agua y mL de
diclorofluoresceína SR. Mezclar, y titular con nitrato de plata SV 0.1N hasta
que flocule el cloruro de plata y la mezcla adquiera color rosa débil. Cada mL
de nitrato de plata 0.1N es equivalente a 3.545 mg de Cl-
E-2.8. SOLUCIÓN DE RINGER LACTATO INYECTABLE.
La solución de Ringer Lactato
Inyectable es una solución estéril de Cloruro de Calcio, Cloruro de Potasio,
Cloruro de Sodio y Lactato de Sodio en Agua para Inyectables. Contiene, no
menos del 95%, y no más del 105% de la cantidad rotulada de ClNa, no menos del
90% y no más del 110% de la cantidad rotulada de CIK, no menos del 90% y no más
del 110% de la cantidad rotulada de Cl2Ca2H2O
y no menos del 90% y no más del 110% de la cantidad rotulada de C3H5NaO3.
La Solución de Ringer Lactato Inyectable no contiene agentes antimicrobianos.
E-2.8.1. Especificaciones y procedimientos de Control
E-2.8.1.1. Envasado y Conservación:
Conservar en envases monodosis de plástico o vidrio, éste último preferentemente
de tipo I o tipo II.
E-2.8.1.2. Rotulado. El rótulo responderá
a lo especificado en el Reglamento Técnico para SPGV punto 8.
E-2.8.1.3. Identificación. Responde a los
ensayos para Sodio (USP XXII-191), para Cloruro (USP XXII-191) y cuando está concentrada
a la mitad de su volumen original, al ensayo de Calcio (USP XXII-191) y al
ensayo de llama para Potasio (USP XXII-191).
E-2.8.1.4. Sustancias piretógenas. Podrá
optarse por uno de los siguientes ensayos:
a) Ensayo de piretógenos: Cumple con los
requerimientos del ensayo de piretógenos (Anexo L.2).
b) Ensayo de endotoxinas bacterianas: Se llevará a
cabo de acuerdo a lo descripto en "Ensayo de endotoxinas bacterianas"
(Anexo L.3). No deberá contener más de 0,5 UE/mL.
E-2.8.1.5. pH (USP XXII-791). Entre 6.0 y 7.5.
E-2.8.1.6. Metales Pesados (USP
XXII-231). Evaporar 67 mL a un volumen de 20 mL, agregar 2 mL de ácido acético
1N, luego diluir con agua a 25 mL: el límite es de 0.3 ppm.
E-2.8.1.7. Otros requerimientos. Cumple
con los "Requerimientos generales para las SPGV" (E.3.1).
E-2.8.1.8. Valoración de Calcio. Proceder
con la Solución de Ringer Lactato Inyectable como se indica en la valoración de
Calcio de la Solución de Ringer Inyectable
E-2.8.1.9. Valoración de Potasio.
Proceder con la Solución de Ringer Lactato Inyectable como se indica en la
valoración de Potasio de la Solución de Ringer Inyectable.
E-2.8.1.10. Valoración de Sodio.
Proceder con la Solución de Ringer Lactato Inyectable como se indica en la valoración de Sodio de la Solución de Ringer Inyectable.
E-2.8.1.11. Valoración de Cloruro.
Proceder con la Solución de Ringer Lactato Inyectable como se indica en la valoración de Cloruro de la Solución de Ringer Inyectable.
E-2.8.1.12. Valoración de Lactato.
Evaporar 50.0 mL de Solución de Ringer
Lactato Inyectable en un crisol o placa adecuada, y quemar suavemente hasta que
esté completamente carbonizado. Separar la masa quemada bien con una varilla de
vidrio, agregar 25 mL de agua y 25,0 mL de ácido sulfúrico 0.1 N SV, y calentar
en baño de vapor durante 30 minutos, separando cualquier grumo con una varilla
de vidrio durante el calentamiento. Filtrar, lavar bien con agua caliente hasta
que el último lavado sea neutro al papel de tornasol, luego enfriar el filtrado
y lavados combinados, agregar naranja de metilo SR, y titular el exceso de
ácido con hidróxido de sodio 0.1 N SV. Cada mL de ácido sulfúrico 0.1 N es equivalente a
8.907 mg de C3H5O3.
E-3. REQUERIMIENTOS GENERALES
E-3.1. Requerimientos generales para las soluciones
parenterales de gran volumen
E-3.1.1. Generalidades
Deben extremarse todos los cuidados en
la preparación de todas las SPGV, para evitar contaminación con microorganismos
y materiales extraños
Las buenas prácticas farmacéuticas
requieren también que cada recipiente final de SPGV sea sometido
individualmente a una inspección física, siempre que la naturaleza del
recipiente lo permita, y que se rechace cada recipiente cuyo contenido muestre
evidencia de contaminación con material extraño visible
E-3.1.2. Agua
El Agua como materia prima para
las Soluciones Parenterales de Gran Volumen debe ser el Agua Para
Inyectables, que responda a la definición y especificaciones establecidas
en el Anexo B punto B.
.
E-3.1.3. Sustancias agregada
Las Soluciones Parenterales de Gran
Volumen no deberán
contener agentes antimicrobianos ni colorantes
Las Soluciones Parenterales de Gran
Volumen no deberán
contener sustancias estabilizantes a menos que estén especificadas en la
monografía correspondiente
3.1.4. Volumen del envase.
Los envases de Soluciones
Parenterales de Gran Volumen deberán contener un ligero exceso no inferior
al 2% del volumen rotulado.
La medida del contenido del envase se
efectuará volcando el mismo en un recipiente calibrado y con graduación
adecuada.
En aquellos envases en que no escurra
sin ventilación, por lo menos el 90% del volumen nominal, deberá agregarse al
rótulo la siguiente advertencia: "Debe administrarse en forma aséptica con
filtración del aire de ventilación".
E-3.1.5. Partículas extrañas.
Todas las Soluciones Parenterales
de Gran Volumen para infusión monodosis deben cumplir con los límites de Partículas
Extrañas establecido en el Ensayo de Partículas Extrañas (E.3.1.13).
Todas las unidades de Soluciones
Parenterales de Gran Volumen deben estar libres de partículas que puedan
ser observadas en una inspección visual a ojo desnudo.
NOTA: Las Soluciones Parenterales de Gran Volumen
envasadas y rotuladas para uso como soluciones para irrigación están exentos de
los requerimientos del ensayo de partículas extrañas.
E-3.1.6. Ensayos de esterilidad.
Las Soluciones Parenterales de Gran
Volumen deben cumplir los requerimientos establecidos en Ensayo de
Esterilidad (Anexo L.4).
E-3.1.7. Rotulado.
Los rótulos deberán responder a los
requisitos generales establecidos en el Documento A-1/91 Reglamento Técnico Soluciones
Parenterales de Gran Volumen ítem 8.
El envase debe rotularse de forma tal
que un área de su longitud o circunferencia total quede sin cubrir para
permitir la inspección del contenido.
NOTA: Los recipientes con Soluciones Parenterales de
Gran Volumen para diálisis peritoneal e irrigación, se deben rotular
indicando que el contenido no es para uso por infusión intravenosa.
E-3.1.8. Envasado y conservación.
En ningún caso las Soluciones
Parenterales de Gran Volumen para infusión endovenosa deberán permitir la
administración de volúmenes mayores de 1 litro.
Las Soluciones Parenterales de Gran
Volumen para irrigación, diálisis o nutrición parenteral están exentas de
la restricción de 1 litro de los requerimientos anteriores en relación al
envasado.
E-3.1.9. Esterilización y seguridad de esterilidad
E-3.1.9.1. Introducción
Dentro de la definición estricta de
esterilidad, una muestra debe considerarse estéril sólo cuando hay ausencia
completa de microorganismos viables en ella. Sin embargo, esta definición
absoluta no puede aplicarse corrientemente a un lote entero de producto
terminado debido a las limitaciones en el ensayo.
Una esterilidad absoluta no puede ser
prácticamente demostrada sin una destrucción completa de cada producto
terminado. La esterilidad de un lote supuesto como estéril se define por lo
tanto en términos probabilísticos, donde la posibilidad de encontrar una unidad
de producto contaminado es aceptablemente remota. Tal estado de seguridad de
esterilidad sólo puede establecerse a través del uso de ciclos de
esterilización adecuados y subsecuente procesamiento aséptico, si existe, bajo
adecuadas buenas prácticas de manufactura, y no confiando solamente en el
ensayo de esterilidad. Los principios básicos para validación y certificación
de un proceso de esterilización se enumeran a continuación:
1) Establecer que el equipo para el proceso tiene
capacidad de operar dentro de los parámetros requeridos.
2) Demostrar que el equipo e instrumentación críticos
para el control son capaces de operar dentro de los parámetros establecidos
para el equipo del proceso.
3) Realizar ciclos repetidos que representen el rango
operacional requerido del equipo, empleando un producto real o simulado.
Demostrar que los procesos han sido realizados dentro de los límites
establecidos en el protocolo y finalmente que la probabilidad de sobrevida
microbiana en los procesos, repetidos completos, no sea mayor que los límites
establecidos.
4) Monitorear el proceso validado durante la
operación de rutina. Periódicamente según se necesite, recalificar y
recertificar los equipos e instrumentos.
5) Completar los protocolos, y documentar los pasos
anteriores.
Para cumplir con los límites
corrientemente aceptables y alcanzables en los parámetros de esterilización, es
necesario emplear instrumentación y equipos adecuados para controlar los
parámetros críticos como temperatura y tiempo. Un aspecto importante del
programa de validación en muchos procedimientos de esterilización implica el
empleo de indicadores biológicos. El proceso validado y certificado debería
revalidarse periódicamente, sin embargo, el programa de revalidación no
necesita necesariamente ser tan extenso como el programa original.
A continuación se detalla un programa
típico para un autoclave de vapor.
El paso de calificación de la
instalación se realiza para establecer que los controles y otra instrumentación
sean adecuadamente diseñados y calibrados. Debe archivarse la documentación
demostrando la calidad de los insumos, como vapor, agua y aire. El paso de calificación
de las operaciones tiene por objeto confirmar que la cámara vacía funciona
dentro de los parámetros de temperatura en todos los lugares claves de la
cámara indicados en el protocolo.
Generalmente es apropiado desarrollar
registros de perfil de calor, por ejemplo, temperaturas simultáneas en la
cámara empleando múltiples sensores de temperatura. Un rango aceptable típico
de temperatura en la cámara vacía es de +/- 1ºC cuando la temperatura de la cámara es no menor a 121 ºC. El paso confirmatorio del programa de validación
es la esterilización real de los materiales o productos. Esta determinación
requiere el empleo de sensores de temperatura dentro de las muestras de los
productos y una de las siguientes alternativas: muestras de los productos a los
cuales se agregaron concentraciones adecuadas de microorganismos de prueba
apropiados o bien indicadores biológicos sueltos en una cámara de autoclave
completamente cargado, con la configuración operacional.
La efectividad de la liberación del
calor o penetración dentro de los productos reales y el tiempo de exposición
son los dos factores principales que determinan la letalidad del proceso de
esterilización. El paso final del programa de validación requiere la
documentación de los datos de apoyo desarrollados al ejecutar el programa.
Generalmente se acepta que los
productos inyectables con esterilización final o dispositivos críticos que se
pretende que sean estériles, cuando se procesan en autoclave, alcanzan una
probabilidad de sobrevida de 10-6, es decir, la seguridad de que la
probabilidad de encontrar microorganismos viables en el producto esterilizado
es menor que uno en un millón. Con productos termoestables, la aproximación
generalmente es a exceder considerablemente el tiempo crítico necesario para
alcanzar una probabilidad de sobrevida microbiana de 10-6
(Overkill).
Sin embargo, con un producto donde una
prolongada exposición al calor puede tener un efecto perjudicial, puede no ser
factible emplear este enfoque.
En este último caso, el desarrollo del
ciclo de esterilización depende mucho del conocimiento de la carga microbiana
del producto basado en el examen, en un período de tiempo adecuado, de un
número considerable de lotes del producto preesterilizado.
Nota: Para el desarrollo y validación de los ciclos de
esterilización por vapor referirse al Anexo H "Validación de ciclos
de esterilización por vapor" de este documento.
E-3.1.9.2. Ensayo de esterilidad de lotes.
Debe reconocerse que el ensayo de
esterilidad de referencia, podría no detectar contaminación microbiana si ésta
está presente sólo en un pequeño porcentaje de los productos terminados en el
lote, debido a que el número especificado de unidades a tomar impone un límete
estadístico significativo sobre la utilidad de los resultados del ensayo. Sin
embargo, esta limitación intrínseca debe ser aceptada ya que el conocimiento corriente
no ofrece alternativas no destructivas para averiguar la calidad microbiológica
de cada producto terminado en el lote, y no es una opción factible aumentar el
número de muestras significativamente.
Los medios principales que apoyan que
un lote de producto terminado supuestamente estéril cumple las
especificaciones, consisten en la documentación de la producción real, el
registro de esterilización del lote y los registros de validación adicionales
que aseguran que el proceso de esterilización posee la capacidad de inactivar
totalmente la carga microbiana establecida en el producto o un desafío
microbiológico mayor.
Si se considera que los datos
derivados de los estudios de validación de la seguridad de esterilidad del
proceso y de los controles del proceso suministran mayor seguridad de que el
lote alcance la baja probabilidad requerida de contener una unidad contaminada
(comparado con los resultados del ensayo de esterilidad de las unidades
terminadas muestreadas de cada lote), cualquier procedimiento de ensayo de
esterilidad adoptado puede ser mínimo. El ensayo de esterilidad generalmente se
realiza directamente después de la manufactura del lote como un ensayo de
calidad final del producto. Los ensayos de esterilidad empleados en esta forma
en el control de manufactura no deben confundirse con los descriptos en el Ensayo
de Esterilidad (L. 4). Los detalles del procedimiento pueden ser los mismos
respecto de los medios, inóculos y manipuleo de las muestras, pero el número de
unidades y/o tiempo(s) de incubación elegidos para el ensayo pueden diferir. El
número debe elegirse en relación al propósito a servir, por ejemplo, de acuerdo
a si se otorga mayor o menor seguridad en el ensayo de esterilidad en el
contexto de todas las medidas para seguridad de esterilidad en la manufactura.
También, tiempos más largos en la incubación harían al ensayo más sensible para
los microorganismos de crecimiento lento. En los ensayos de promoción del
crecimiento para los medios, los microorganismos de crecimiento lento,
particularmente si son aislados de la carga microbiana del producto, deberían
incluirse con las otras cepas de ensayo. Los resultados de esterilidad
negativos o satisfactorios sirven sólo como un aporte más de la evidencia
existente respecto de la calidad del lote si todos los registros de producción
correspondientes del lote están en orden y se sabe que el proceso de
esterilización es efectivo.
E-3.1.9.3. Realización, observación e
interpretación.
Las condiciones para el ensayo de
esterilidad deben ser tales que no ofrezcan un mayor desafío microbiano a los
productos que se analizan que el de un área de producción para procesamiento
aséptico. El procedimiento para el ensayo de esterilidad debería ser realizado
por individuos que tengan un alto nivel de capacitación en técnicas asépticas.
Las grandes manipulaciones asépticas requeridas para realizar un ensayo de
esterilidad pueden resultar en una probabilidad de contaminación no relacionada
con el producto, del orden de 10-3, un nivel similar a la eficiencia
general de una operación aséptica y comparable a la probabilidad de sobrevida
microbiana de los productos asépticamente procesados. Este nivel de
probabilidad es significativamente mayor que el generalmente atribuido a un
proceso de esterilización terminal, una probabilidad de uno en un millón o 10-6
de sobrevida microbiana. Se deberían emplear periódicamente productos
terminados reconocidamente estériles, deberían emplearse periódicamente como
controles negativos, para asegurar la confiabilidad del procedimiento de
ensayo. Preferiblemente los técnico que realizan el ensayo deberían desconocer
que están analizando controles negativos. De estos ensayos es deseable una
frecuencia de falsos positivos que no exceda el 2%.
Sin embargo para los productos efectivamente
esterilizados terminalmente, la menor probabilidad de sobrevida microbiana
puede indicar el uso de un ensayo menos extenso que el procedimiento
especificado en Ensayo de esterilidad. Esta confiabilidad agregada de la
seguridad de esterilidad de la esterilización terminal depende de un proceso de
esterilización validado y documentado.
El ensayo de esterilidad sólo, no es
un substituto.
E-3.1.9.10. Ensayo de partículas extrañas
E-3.1.10.1. Generalidades:
Partículas extrañas son sustancias móviles,
insolubles, diferentes a burbujas de gas, presentes no intencionalmente en las
soluciones parenterales.
Las soluciones parenterales de gran
volumen deben estar libres de partículas que puedan observarse en la inspección
visual. En los siguientes ensayos, para inyectables de gran volumen, los
resultados obtenidos al examinar una unidad aislada o un grupo de unidades no
pueden extrapolarse con certeza a otras unidades que no se han analizado.
Deben elaborarse planes de muestreo
estadísticamente válidos, basados en un grupo conocido de factores
operacionales dados si se quieren obtener deducciones válidas de los datos
observados para caracterizar el nivel de Partículas Extrañas en un gran grupos
de unidades.
E-3.1.10.2. Ensayo.
Este ensayo para partículas extrañas
es adecuado para revelar la presencia de partículas cuyo eje longitudinal, o
dimensión lineal efectiva, es de 10 Om o superior. Pueden emplearse
procedimientos alternativos para medir partículas, siempre que los resultados
obtenidos sean de confiabilidad equivalente.
Sin embargo, cuando aparece una
diferencia, o en el caso de una duda, sólo es concluyente el resultado obtenido
por el procedimiento dado en esta Norma.
E-3.1.10.3. Procedimiento.
Nota: En todo este procedimiento, usar guantes
adecuados sin polvo y material de vidrio y equipos escrupulosamente limpios que
hayan sido enjuagados sucesivamente con una solución caliente de detergente,
agua caliente, agua y alcohol isopropílico. Aplicar el agua como un chorro de
atrás hacia adelante a través de la superficie del objeto sostenido
verticalmente, trabajando lentamente de arriba hacia abajo. Realizar el
enjuague con alcohol isopropílico bajo una campana de flujo laminar equipada
con filtros ultra-HEPA (aire particulado de alta eficiencia). Dejar secar los
objetos bajo la campana a contracorriente de las demás operaciones.
Preferiblemente, colocar la campana en una habitación separada con aire
acondicionado, filtrado y mantenida bajo presión positiva respecto del área
circundante. Antes de concluir el ensayo, limpiar la campana de flujo laminar
(excepto las superficies de los medios de filtro) con un solvente apropiado.
Mantener una velocidad de flujo de aire a 90 - 20 pies/minuto).
E-3.1.10.4. Equipo y membrana filtrante.
Usando pinzas, retirar una membrana
reticulada de color contrastante de su envase. Lavar ambos lados de la membrana
con una corriente de agua que ha sido filtrada además a través de una membrana
adecuada para eliminar partículas que tengan una dimensión efectiva lineal
mayor de 5 um, sosteniendo el filtro en posición vertical, y comenzando por la
parte superior del lado sin reticulado, pasando la corriente de atrás hacia
adelante a través de la superficie, trabajando lentamente desde arriba hacia
abajo de forma que las partículas se enjuaguen bajando hacia el filtro, y
repitiendo el proceso sobre el lado reticulado. Colocar la membrana (el lado
reticulado hacia arriba) sobre a base del porta filtro, e instalar el embudo de
filtración sobre la base sin deslizar el embudo sobre el filtro de membrana.
Invertir la unidad armada, y lavar el interior del embudo aproximadamente 10
segundos con un chorro de agua filtrada. Dejar drenar el agua, y colocar la
unidad sobre el recipiente de filtrado.
E-3.1.10.5. Realización.
Mezclar la solución invirtiendo el
envase 20 veces. Limpiar profundamente la superficie externa del envase con un
chorro de agua filtrada, y retirar el cierre cuidadosamente evitando la
contaminación del contenido. Transferir 25 mL de la solución bien mezclada al
embudo, dejar descansar 1 minuto, y aplicar vacío y filtrar. Retirar el vacío
suavemente, y lavar las paredes interiores del embudo con un chorro de 25 mL de
agua filtrada. Dirigir el chorro de agua filtrada de forma tal de lavar las
paredes del embudo dejándolas sin partículas que puedan quedarse sobre las
paredes, pero evitando dirigir la corriente sobre la superficie del filtro.
Después que desapareció la turbulencia, filtrar con vacío el enjuague. Retirar
suavemente la sección superior del equipo filtrante mientras se mantiene el
vacío, interrumpir el vacío, y retirar con pinzas la membrana. Fijar la
membrana en una placa de petri, usando una película muy delgada de grasa de
robinete, si es necesario para mantener el filtro plano y en su lugar. Dejar
secar la membrana con la caja de petri semicerrada. Colocar cuidadosamente la
placa de petri sobre la platina del microscopio, y contar las partículas sobre
el filtro como se describe más adelante.
E-3.1.10.6. Determinación.
Examinar la membrana completa en un
microscopio adecuado con un aumento de 100 X con la luz incidente en un ángulo
de 10º a 20º con la horizontal. Contar el número de partículas que tengan una
dimensión lineal efectiva igual o mayor que 10 Om e igual o mayor que 25 Om.
Realizar la determinación en un blanco, usando un equipo y membrana filtrante
como se indica en Realización del ensayo, comenzando con "lavar las
paredes interna del embudo con un chorro". Restar el recuento total
obtenido en el blanco al recuento total no corregido obtenido en la muestra.
Nota: Para las soluciones que contienen dextrosa, no
enumerar material morfológicamente indistinto que muestre poco o ningún relieve
de superficie y que presente un aspecto gelatinoso o tipo película. Como en
solución este material consiste en unidades del orden de 1 Om o menos y es
probable que sea contado sólo después de su agregación o deformación de la
membrana, la interpretación de su recuento puede ayudarse analizando una
muestra de la solución con un contador electrónico de partículas adecuado.
E-3.1.10.7. Interpretación.
Examinar muestras y blanco por
duplicado como se indicó. Si la determinación del blanco da más de 5 partículas
que tienen dimensión lineal efectiva de 25 Om o más, el entorno operacional es
no satisfactorio y el ensayo no es válido.
Los Inyectables de gran volumen para
infusión monodosis cumplen con los requerimientos del ensayo si contienen no
más de 50 partículas por mL que sean igual o mayores de 10 Om y no más de 5
partículas por ml que sean iguales o mayores de 25 Om en su dimensión lineal
efectiva.
E-4. REACTIVOS
E-4.1. Soluciones SR.
Ver características, preparación y
usos en: "Test Solutions (TS)" en USP XXII páginas 1786-1792.
E-4.2. Soluciones SV.
Ver características, preparación y
usos en: "Volumetric Solutions (VS)" en USP XXII páginas
1792-1798.
ANEXO F
TRANSPORTE DE SOLUCIONES PARENTERALES
DE GRAN VOLUMEN
CONTENIDO
F-1
OBJETIVO
F-2
DEFINICIONES
F-3
CONDICIONES GENERALES
F-1 OBJETIVO
Esta norma establece los procedimientos a ser observados
a fin de evitar que las soluciones parenterales de gran volumen (SPGV) sufran
alteraciones durante su transporte. En la distribución a nivel primario, el
cumplimiento de esta norma es responsabilidad del fabricante.
F-2. DEFINICIONES
F-2.1. Soluciones parenterales de gran volumen (SPGV).
Son soluciones en base acuosa,
estériles, apirógenas, acondicionadas en un recipiente único de 100 mL o mayor
y esterilizadas terminalmente. Se incluye en esta definición tanto las
soluciones para administración endovenosa cuando las destinadas a la irrigación
o a la diálisis peritoneal. El término "parenteral de gran volumen"
no incluye ningún producto de origen biológico.
F-2.2. Fabricante.
Persona jurídica que elabora SPGV, con
la previa autorización de funcionamiento por parte de la autoridad sanitaria
nacional competente.
F-2.3. Distribuidor.
Persona jurídica que realiza fases de
comercialización de SPGV
F-2.3.1. Distribuidor a nivel
primario.
El que entrega en forma directa en la
cadena de comercialización, promoción e investigación aplicada, desde el
fabricante del producto hasta el primer receptor del mismo.
F-2.4. Transportador.
La empresa que realiza el transporte
de SPGV (en caja cerrada)
F-3. CONDICIONES GENERALES
F-3.1. E transporte de SPGV debe
hacerse de manera tal que no se afecte la identidad, integridad o pureza de las
mismas.
F-3.2. La empresa transportadora debe
ofrecer condiciones que garanticen la ejecución de ese servicio, en un todo de
conformidad con la presente norma.
F-3.3. La persona responsable del
transporte debe ser debidamente orientada y entrenada para seguir las
indicaciones de esta Norma.
F-3.4. La carga, transporte, descarga
y almacenamiento de los productos debe seguir las siguientes recomendaciones:
F-3.4.1. Los
vehículos o depósitos deben estar perfectamente limpios y exento de cualquier
suciedad u olor.
F-3.4.2. No
se deben transportar o depositar los productos en ambientes húmedos, sin
ventilación o expuestos al sol.
F-3.4.3. Las
SPGV deben ser transportadas y depositadas bajo condiciones tales de seguridad
que aseguren que no se afecte su integridad y calidad. En especial, no deben
ser transportadas con los productos que se enumeran a continuación:
a) alimentos
y materiales perecederos;
b) disolventes
orgánicos;
c) gases;
d) sustancias
corrosivas y/o tóxicas;
e)
pesticidas, agrotóxicos;
f) materiales
radiactivos.
F-3.4.4.
Respetar el apilado máximo recomendado por el fabricante.
F-3.4.5.
Apilar los productos de acuerdo con los símbolos presentes en los embalajes.
F-3.4.6. Se
debe tener cuidado con los embalajes durante el transporte o almacenamiento de
los productos. Evitar juegos, utilizarlos como asiento y caminar sobre los
mismos a fin de no dañarlos.
F-3.4.7.
Proteger las cajas de la intemperie (lluvia, sol) y del ataque de insectos y
roedores.
F-3.5.
Cualquier sospecha de daños en los productos debe ser comunicada inmediatamente
al fabricante a fin de que se tomen las providencias necesarias.
F-3.6. La
entrega del material debe realizarse en presencia de una persona debidamente
autorizada por el establecimiento para la recepción del producto.
F-3.7. En
caso de siniestro el transportador debe comunicarlo inmediatamente al
fabricante a fin de que se tomen las providencias necesarias.
ANEXO G
SOLUCIONES PARENTERALES DE GRAN VOLUMEN RECEPCION,
ALMACENAMIENTO Y DISPENSACION
CONTENIDO
G-1 OBJETIVO
G-2
DEFINICIONES
G-3
CONDICIONES GENERALES
G-1. OBJETIVO
Esta Norma establece los procedimientos de
recepción, almacenamiento y dispensación de soluciones parenterales de gran
volumen, con el objeto de evitar que las mismas sufran alteraciones durante
tales operaciones.
G-2. DEFINICIONES
G-2.1. Solución parenteral de gran volumen (SPGV).
Son soluciones en base acuosa,
estériles, apirógenas, acondicionadas en un recipiente único de 100 mL o mayor
y esterilizadas terminalmente. Se incluye en esta definición tanto las
soluciones para administración endovenosa cuando las destinadas a la irrigación
o a la diálisis peritoneal. El término "parenteral de gran volumen"
no incluye ningún producto de origen biológico.
G-2.2. Fabricante.
Persona jurídica que elabora SPGV, con
la previa autorización de funcionamiento por parte de la autoridad sanitaria
nacional competente.
G-2.3. Distribuidor.
Persona jurídica que realiza fases de
comercialización de SPGV
G-2.4. Transportador:
La empresa que realiza el transporte
de SPGV (en caja cerrada)
G-2.5. Lote.
Conjunto de SPGV que se produce en un
ciclo de fabricación, cuya característica esencial es la homogeneidad.
G-2.6. Número de lote.
Designación impresa en el rótulo de
cada unidad del producto, constituida por combinaciones de letras, números o
símbolos, que permita identificar el lote a que ésta pertenece y, en caso de
necesidad, localizar y repasar todas las operaciones de producción, inspección
y control practicadas durante la fabricación, acondicionado, almacenamiento y
distribución del mismo.
G-2.7. Cuarentena.
Retención temporaria de un lote de
producto con la prohibición de usarlo hasta que el mismo sea aprobado por el
Control de Calidad.
G-3. CONDICIONES GENERALES
G-3.1. Recepción
G-3.1.1. La recepción de SPGV debe estar orientada por
procedimientos escritos que incluyan directivas específicas con respecto a cada
producto, en un todo de acuerdo a las recomendaciones de esta Norma.
G-3.1.2. La recepción debe ser efectuada por personal
debidamente habilitado y entrenado en cuanto a las características del producto
de manera de evaluar sus condiciones.
G-3.1.3. El responsable de la recepción debe consignar y
anotar en a factura:
a) nombre de (los) producto(s);
b) nombre del fabricante;
c) número de lote;
d) nombre del transportador;
e) número de patente del vehículo;
f) tipo de vehículo (cerrado, abierto con cubierta,
furgón, etc.);
g) condiciones higiénicas;
h) condiciones de estibado de la carga;
i) fecha y hora de llegada.
G-3.1.4. Se deberán tener en cuenta las siguientes
observaciones en la descarga de material:
a) evitar golpes que puedan ocasionar daños al
producto;
b) verificar y separar los productos de acuerdo con
sus números de lote, para facilitar su almacenamiento;
c) inspeccionar visualmente algunas unidades para
verificar la integridad de las mismas.
G-3.1.5. En caso de que el vehículo sea considerado
inadecuado o bien que los productos presentaran daños en su embalaje externo,
la carga debe ser puesta en cuarentena debidamente identificada y el comprador
deberá comunicar por escrito lo ocurrido al fabricante o distribuidor y enviar
copia a la autoridad sanitaria si lo considerase necesario.
G-3.2. Almacenamiento
G-3.2.1. El almacenamiento de SPGV debe estar orientado por
procedimientos escritos que incluyan indicaciones específicas para cada
producto de acuerdo con las recomendaciones de esta Norma.
G-3.2.2. El lugar de almacenamiento debe tener capacidad
suficiente para permitir la separación selectiva y ordenada de los productos y
la rotación de stocks.
G-3.2.3. El área de almacenamiento debe estar seca,
ventilada, protegida del sol y limpia.
G-3.2.4. El almacenamiento de los productos se debe
realizar en condiciones adecuadas de temperatura, humedad e iluminación, de
acuerdo con las instrucciones del fabricante, de manera de no afectar la
identidad y calidad del producto.
G-3.2.5. El apilado de las cajas debe hacerse separado del
suelo para facilitar la limpieza, y seguir las instrucciones del fabricante en
cuanto al máximo de cajas a apilar.
G-3.2.6. El almacenamiento debe ser ordenado de manera que
permita individualizar cada lote y dispensar los mismos en orden cronológico de
sus fechas de vencimiento.
G-3.3.
Dispensación
G-3.3.1. La dispensación de SPGV debe ser orientada por
procedimientos escritos que incluyan instrucciones específicas para cada
producto conforme a las recomendaciones de esta Norma.
G-3.3.2. Antes de la dispensación se debe:
a) identificar el número de lote y su fecha de
vencimiento;
b) inspeccionar visualmente cada unidad a ser
dispensada y verificar el aspecto de la solución y la integridad del recipiente;
c) transportar el material en forma adecuada,
evitando comprometer el embalaje y sin retirar la protección plástica o de
cartón;
d) llevar un registro de dispensación por lote y área
de uso.
En caso de que desde el área de uso se
comuniquen observaciones de reacciones adversas u otras, separar el lote y
comunicar inmediatamente, por escrito, al fabricante o distribuidor y a la
autoridad sanitaria si se considera necesario.
ANEXO H
VALIDACION DEL PROCESO DE
ESTERILIZACION POR VAPOR
CONTENIDO
H.1
OBJETIVO
H.2
DEFINICIONES
H.3
CONDICIONES GENERALES
H.4
CONDICIONES ESPECÍFICAS
H.5
CRITERIOS PARA LA REVALIDACIÓN
H-6
RECERTIFICACIÓN
H-1.
OBJETIVO
Esta norma establece las condiciones exigidas para
validar un ciclo de esterilización por calor húmedo, de modo de garantizar la
eficiencia del proceso, es decir, garantizar una probabilidad de sobrevida
microbiana no superior a 1 x 10-6 (menos de una unidad no estéril
por cada millón de unidades) para productos parenterales esterilizados
terminalmente.
H-2.
DEFINICIONES
A efecto de ésta norma, son adoptadas las
siguientes definiciones:
H-2.1. Validación. Programa formal para comprobar
la eficiencia y reproducibilidad de una técnica, operación o proceso.
H-2.2. Validación de ciclos de
esterilización. Procedimiento
que confirma que la letalidad del ciclo es suficiente para garantizar una
probabilidad de sobrevida microbiana no superior a 1 x 10-6.
H-2.3. Protocolo. Documento conteniendo una descripción
del programa a seguir en la evaluación del proceso de esterilización.
H-2.4. Certificación. Función administrativa en la cual se
realiza la revisión y la aprobación del proceso, como etapa final del programa
de validación.
H-2.5. Esterilización terminal. Procedimiento
aplicado a recipientes cerrados que contienen SPGV, garantizando una
probabilidad de sobrevida microbiana no superior a 1 x 10-6 en el
producto.
H-2.6. Esterilidad. Es la ausencia de microorganismos
viables. Se supone que los productos que cumplen los criterios de
esterilización terminal están estériles.
H-2.7. Biocarga pre-esterilización. Número e
microorganismos viables presentes en el producto, envasado y cerrado, antes de
la esterilización.
H-2.8. Indicador biológico. Es un sistema conteniendo microorganismos de concentración y resistencia
térmica conocidas, del cual se puede anticipar una tasa de mortalidad
previsible cuando es expuesto a parámetros físicos específicos.
H-2.9. Calificación. Parte del programa de validación, donde el control
de los parámetros físicos del sistema de esterilización es evaluado para
demostrar su adecuación para realizar lo que fue propuesto en el proceso
proyectado.
H-2.10. Recalificación. Repetición de una parte o de todos
los requisitos de la Calificación para reevaluar la utilidad del proceso.
H-2.11. Estudio de distribución de
calor. Estudio para
documentar que la distribución del medio esterilizante en el interior de la
cámara garantiza que todos los recipientes reciban una cantidad aceptable y
uniforme de calor, durante el proceso de esterilización.
H-2.12. Estudio de penetración de calor. Estudio
para asegurar que el recipiente menos calentado en el interior de la carga sea
suficientemente expuesto al calor letal.
H-2.13.
Valor "D". Tiempo,
expresado en minutos, a determinada temperatura, necesario para conseguir una
reducción logarítmica (o del 90%) en el número de microorganismos.
H-2.14.
Valor "z". Número
de grados de temperatura necesarios, bajo condiciones específicas, para
conseguir una reducción logarítmica (o del 90%) en el valor "D".
H-2.15. Valor de "Fzt". Tiempo equivalente a una temperatura t suministrado a un
producto con el propósito de su esterilización, para un valor específico de z.
H-2.16. Valor de Fo: Tiempo equivalente
a 121 "C suministrado a un producto con el propósito de su esterilización,
para un valor de z = 10.
H-3. CONDICIONES GENERALES
H-3.1. Este documento fija los procedimientos a seguir
para la calificación y validación del proceso de esterilización de las SPGV, de
modo de garantizar su eficiencia.
H-3.2. La esterilización requiere un abordaje
multidisciplinario, integrando programas microbiológicos, físicos y de
ingeniería, para garantizar la esterilidad, sin comprometer la calidad total
del producto.
Características de las solucione tales
como potencia, composición química, pH, ausencia de partículas, no deben ser
adversamente afectadas por el proceso de esterilización.
H-3.3. Una SPGV está definida como estéril si sufrió una
esterilización terminal. La eficiencia de la esterilización terminal puede
establecerse por el uso de indicadores biológicos apropiados y métodos físicos
que relacionen la resistencia térmica de la biocarga de pre-esterilización al
ciclo de esterilización terminal. En este caso, la resistencia al calor y la
biocarga de pre-esterilización deben ser conocidas y monitoreadas.
H-4. CONDICIONES ESPECIFICAS
H-4.1. Estudios de pre-validación
Son un grupo de actividades
pre-establecidas en procedimientos escritos, que deben ser completadas y
documentadas antes de que se inicie la validación del ciclo de esterilización.
Los estudios de pre-validación involucran:
H-4.1.1. Definición de los parámetros físicos del ciclo,
los criterios de control y los límites de tolerancia. Estos límites deben ser
establecidos para garantizar que se alcancen los requisitos mínimos para la
letalidad, y que no puedan ocurrir efectos adversos sobre el producto.
H-4.1.2. Documentación adecuada de cada operación.
H-4.1.3. Calibración, contra patrones certificados, de
todos los equipos e instrumentos de medición usados en la pre-validación.
H-4.1.4. Determinación de las características del producto
que puedan ser afectadas por influencia del proceso de esterilización, tales
como:
a) integridad física del recipiente de SPGV durante y
después del proceso.
b) Evaluaciones microbiológicas del sistema de
cerrado del recipiente.
c) Estabilidad del producto bajo las condiciones de
tiempo y temperatura.
H-4.1.5. Definición de las configuraciones de las cargas y
establecimiento de la densidad de carga para que existan condiciones uniformemente
reproducibles de transferencia de calor.
H-4.2. Validación
H-4.2.1.
Introducción.
La validación de la esterilización es
necesaria en las siguientes situaciones:
a) Para la confirmación de la eficiencia del proceso empleado.
b) Con cada cambio de las condiciones de un ciclo.
c) Al instalar un nuevo equipo.
Deben realizarse estudios de
validación, en los cuales todos los criterios de aceptación descriptos en el
protocolo deben ser cumplimentados.
La validación debe incluir:
a) Toda la documentación de los equipos.
b) La preparación del protocolo.
c) Los ensayos de calificación para demostrar la adecuación del proceso
y, por tanto, la certificación final.
H-4.2.2.
Protocolo de validación:
El protocolo de validación de la
esterilización debe ser preparado con participación de los técnicos
especializados en ingeniería, producción y control de calidad de producto.
El protocolo debe contener:
H-4.2.2.1. Especificaciones e identificaciones del equipo,
producto y proceso de esterilización a ser calificado.
H-4.2.2.2. Especificación del tipo de equipo a
usar para la recolección de datos, método de calibración e intervalos de tiempo
en la recolección de datos.
H-4.2.2.3. Criterios de aceptación para:
a) Ciclo de esterilización;
b) Distribución y penetración de calor;
c) Control y desempeño de indicadores biológicos;
d) Aprobación del proceso de esterilización.
H-4.2.3. Calificación de los equipos y
las instalaciones.
El programa de calificación debe ser
establecido antes del uso rutinario de un sistema de esterilización nuevo o
modificado. La finalidad es la de asegurar que el equipo sea capaz de
reproducir los parámetros dentro de los límites establecidos para las
especificaciones del proceso.
La calificación de los equipos debe incluir la
descripción y/o diseños, proyectos, diagrama de flujos de:
a) Cámara de esterilización;
b) Sistema de cañerías;
c) Sistemas de instrumentación (los instrumentos usados deben ser
calibrados contra patrones certificados).
H-4.2.4. Métodos:
Se reconocen dos procedimientos
básicos para la definición de los ciclos de esterilización en función de la
termorresistencia de los productos a ser esterilizados.
H-4.2.4.1. Método de la probabilidad de sobrevida
a) Fundamentos e indicaciones:
El método de la probabilidad de
sobrevida establece los parámetros del ciclo en base al número de
microorganismos que se encuentran presentes normalmente en el producto a ser
esterilizado y en su resistencia térmica.
La combinación de estos datos
determina la cantidad mínima de calor necesario para reducir la biocarga de
pre-esterilización a una probabilidad de sobrevida microbiana no superior a 1 x
10-6.
Generalmente este criterio se emplea
cuando se desarrollan y se validan ciclos de esterilización para productos
termosensibles. Sin embargo, si uno lo elige, podría ser empleado para
materiales termoestables.
b) Etapas del método de probabilidad de sobrevida:
|
Probabilidad
De sobrevida
|
Estudios de Laboratorio (H-4.3)
Estuidos Industriales
(H-4.4)
|
Estudios de la Biocarga de preesterilización
(H-4.3.1.)
|
|
|
|
|
|
Estudio de la resistencia térmica de la biocarga
(H-4.3.2.)
|
Determinación valor D
(H-4.3.3)
Determinación valor z
(H-4.3.4)
|
|
|
|
|
Cálculo de la probabilidad de sobrevida y la
determinación del valor F mínimo para el ciclo (H-4.3.5)
|
|
|
|
|
|
Estudios de distribución de calor (H-4.4.1)
|
|
|
|
|
|
|
|
Estuidos de penetración de calor (H-4.4.2)
|
|
|
|
|
|
|
|
|
|
Criterios de aceptación de los estudios de
penetración de calor (H-4.4.2.3)
|
|
|
|
|
|
|
|
|
|
Cálculo de F (H-4.4.3)
|
|
|
|
|
|
|
|
|
Biovalidación (H-4.5)
|
|
|
H-4.2.4.2. Método de sobremuerte ("overkill")
a) Fundamentos e indicaciones:
El método de sobremuerte establece por
lo menos la misma probabilidad de sobrevida microbiana (no superior a 10-6, sin
tener en cuenta el número de microorganismos viables -biocarga- y su
resistencia térmica. Este método es ampliamente empleado cuando se esterilizan
materiales termoestables.
b) Etapas del método de sobremuerte:

H-4.3. Estudios de laboratorio
H-4.3.1. Estudios de la biocarga de pre-esterilización.
Consiste en la determinación del número
de microorganismos asociados con el producto. El número y la frecuencia de los
lotes analizados deben ser determinados por el fabricante en base a la
variación del número de microorganismos de lote a lote, en el potencial
crecimiento microbiano antes de la esterilización y en la variación estacional
de la biocarga.
Los recuentos microbianos
(determinación del número de unidades formadoras de colonias-ufc) de SPGV deben
ser realizados en áreas adecuadas para ensayos microbiológicos (flujo laminar),
conforme a la siguiente metodología:
a) Filtrar un volumen adecuado de la SPGV, contenida en el recipiente final, a través de un filtro de membrana de 0.45 micrones
(el método de recuento de colonias en placa puede ser utilizado cuando el
número de microorganismos en la solución sea elevado).
b) Inocular las membranas con un medio de cultivo no
selectivo adecuado (Soybean Casein Digest Agar, Tryptorie Clicose Extract Agar,
Eugon Agar) a 30ºC-35ºC por un mínimo de 72 (setenta y dos) horas.
c) Calcular el número de unidades formadoras de
colonias (ufc) por recipiente de SPGV.
H-4.3.2. Estudio de la resistencia térmica (RT) de los
microorganismos.
En los productos termolábiles, el
estudio de la RT de los microorganismos asociados con el producto y definidos
en el estudio de la biocarga de pre-esterilización es necesario para determinar
el tiempo mínimo de esterilización, para una reducción decimal de a biocarga a
determinada temperatura (valor D), de modo de ofrecer una garantía aceptable
para el proceso. Los microorganismos resistentes deben ser aquellos
periódicamente aislados de varios productos, que por medio de ensayos de
selección demuestran ser resistentes al calor.
H-4.3.2.1. Sistemas para la determinación de la RT de los microorganismos.
Existen diversos tipos de aparatos
para la determinación precisa y reproducible de la RT de los microorganismos. Los más simples son:
a) Baño de aceite o glicerol: Amplias selladas o tubos
capilares conteniendo la suspensión líquida de los microorganismos se sumergen
en el baño de aceite o glicerol a una determinada temperatura, constante, por
un tiempo también determinado.
b) Retorta en miniatura o autoclave: Varios tipos de
muestras (suspensiones en ampollas, tiras de papel inoculadas, esporas
inoculados en vehículos sólidos, o productos inoculados) pueden ser expuestos
dentro de la cámara, que puede suministrar fases rápidas de calentamiento y
enfriamiento durante el período de exposición.
H-4.3.2.2. Ensayos de selección de RT
para la biocarga de un producto.
El objetivo de los ensayos de
selección es el de seleccionar los microorganismos resistentes para los cuales
el valor D debe ser determinado.
a) Dar un choque térmico a la solución, durante 10 a 15 (diez a quince) minutos a 80ºC-100ºC, para eliminar células vegetativas y estimular la
esporulación de los microorganismos.
b) Utilizar la suspensión de esporas resultantes para
la determinación del valor D.
H-4.3.3. Determinación del valor D (reducción
decimal de a biocarga, a determinada temperatura).
H-4.3.3.1. Metodología
a) Con las formas esporuladas obtenidas en los
ensayos de selección, inocular muestras de SPGV, en duplicado, con un número
conocido de esporas o utilizar tiras de papel o vehículos sólidos conteniendo
también un número conocido de esporas.
b) Preparar controles positivos para determinar el
número de esporas en la suspensión (o tira, etc.) a ser analizada. El número de
esporas necesarias será basado en el esquema de cada experimento, pero
generalmente está en el orden de 104 a 107 esporas por muestra.
c) Exponer por los menos 0.5 (cinco) muestras a una
temperatura especificada durante, por lo menos, 3 (tres) intervalos de tiempo
diferentes, para determinar el número de sobrevivientes (Cálculo del valor D).
H-4.3.3.2. Cálculo del valor D.
El valor D o tiempo de reducción
decimal, a una determinada temperatura, es el tiempo necesario para inactivar
el 90% de la población microbiana del producto, y puede ser calculado por
curvas de sobrevida (recuento de ufc) o por el método de fracciones negativas.
a) Cálculo por curvas de sobrevida (recuento de ufc).
a.1) Con los datos de sobrevida, realizar una curva
semilogarítmica, colocando en un gráfico el logaritmo del número de
sobrevivientes (en e eje y) y el tiempo de calentamiento a una determinada temperatura
(en el eje x).
a.2) Ajustar la recta de regresión lineal, siguiendo
una ecuación de
y = a + bx
Considerando:
y log10 del número de sobreviviente en
el tiempo x
x tiempo de calentamiento a una temperatura
determinada
a intersección del eje y a tiempo O
b pendiente de la recta
El valor D es la recíproca negativa de la pendiente
de la recta.
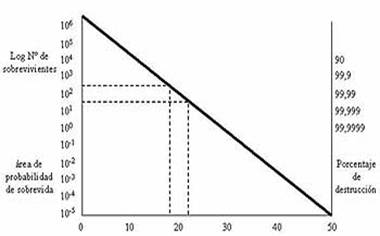
Fig. 1 -
Curva de sobrevida
b) Cálculo por el método de fracciones negativas
b.1) Por lo menos 10 (diez) muestras inoculadas con el
mismo número de esporas son calentadas a una temperatura determinada durante,
por lo menos, 3 (tres) intervalos de tiempo diferentes.
b.2) Luego del calentamiento, las muestras son
incubadas en un medio adecuado (Soybeam Casein Digest Medium, Thioglycolate
Fluid, etc.) a una temperatura ideal para el crecimiento de los
microorganismos, por un mínimo de 7 (siete) días.
b.3) Luego de 7 (siete) días de incubación registrar la
fracción de muestras negativas (sin crecimiento), siendo:
U= tiempo de exposición a la temperatura
especificada.
A= concentración inicial de microorganismos en la
muestra
B=
2,303 log10 (n/q)
N= número total de muestras inoculadas
Q= número total de muestras negativas
luego de la esterilización
U
D=
----------------------------------------
Log10A – log10B
H-4.3.4. Determinación del valor z (número de grados de temperatura
necesarios para obtener una reducción logarítmica del valor D):
El valor z, se define como el número
de grados de temperatura necesario para cambiar el valor D en un factor de 10;
es útil para realizar cálculos que permitan la comparación de la letalidad de
las esporas a diferentes temperaturas.
Para los fines del cálculo, cuando involucre la
resistencia térmica de los microorganismos naturales, es apropiado presuponer
un valor z = 10ºC.
Cuando se utilizan indicadores
biológicos para medir la letalidad durante la validación, se debe verificar el
valor z de los mismos, ya que pueden existir discrepancias entre los valores de
Fo determinados por sensores de temperatura (Fo presupone un z 10ºC) y valores F determinados por indicadores biológicos cuando el valor z de los indicadores varía
significativamente de 10ºC.
H-4.3.5. Cálculo de la probabilidad de sobrevida y del
valor F mínimo necesario para la esterilización: usando los datos de la
biocarga y de los valores D y z. El valor usado para representar la biocarga,
es generalmente, el número máximo de microorganismos (recuento total por
recipiente) encontrado en un producto dado. Cuando existe preocupación por los
efectos adversos de un calentamiento excesivo, es aceptable considerar a la
biocarga como el número máximo de bacterias formadoras de esporas por recipiente
de producto. Los valores D usados en los cálculos del proceso son generalmente
aquellos obtenidos en los microorganismos más resistentes aislados en el
producto, presuponiendo que toda la población esté constituida por los
microorganismos más resistentes al calor.
H-4.3.5.1. Determinación de la
probabilidad de sobrevida.
Cuando se conoce el valor F para un
ciclo de esterilización, la probabilidad de sobrevida microbiana para este
ciclo es calculada a través de la siguiente fórmula:
Log10B = Log10A_ F
D
donde:
B = probabilidad de sobrevida (nivel
máximo aceptable para la probabilidad de sobrevida 1x10-6)
A = biocarga del producto.
D = tiempo necesario para reducir en
un 90% la población de microorganismos más resistentes encontrados en un
producto.
F = letalidad mínima necesaria,
presuponiendo z= 10ºC, expresada como el número de minutos en que el recipiente
más frío en la carga debe ser calentado a la temperatura especificada.
H-4.3.5.2. Determinación del valor F
mínimo necesario.
Cuando se conoce la biocarga de un
producto, la resistencia de los microorganismos naturales y e nivel máximo de
sobrevida microbiana aceptable, F se calcula por la siguiente fórmula:
F = D (log10 A- log10 B)
B = nivel máximo aceptable para la probabilidad de
sobrevida.
H-4.3.5.3. Determinación del tiempo de exposición.
El tiempo de exposición de un ciclo de
esterilización necesario para suministrar el valor F mínimo exigido, puede ser
determinado de la siguiente manera:
Establecer el punto frío de la carga
por sensores de temperatura y ajustar el tiempo de esterilización, de modo que
el recipiente más frío esté a la temperatura de exposición por el tiempo
especificado. Esto no considera la letalidad adicional recibida por el producto
durante las fases de calentamiento y enfriamiento del ciclo de esterilización.
H-4.4. Estudios industriales.
La validación de un nuevo proceso de
esterilización debido a nuevas condiciones del ciclo o nuevos equipos, incluye
estudios de calificación en los cuales se debe cumplir todos los criterios de
aceptación descriptos en el protocolo. Cada cámara de esterilización
(autoclave) de producción debe ser calificada. Cuando una serie de autoclaves
idénticas hayan sido calificadas para un mismo ciclo de esterilización, se
podrán biovalidar nuevos productos en cualquiera de las autoclaves idénticas.
Si un
producto nuevo pudiera ser esterilizado usando un ciclo previamente calificado
por su semejanza un producto ya calificado, el producto nuevo puede ser
calificado por equivalencia sólo para ser esterilizado empleando aquellas
autoclaves calificadas.
H-4.4.1. Estudios de distribución de calor
H-4.4.1.1. Introducción.
Estos estudios se deben realizar en
toda autoclave para cada configuración de carga y para cada tamaño de
recipiente a no ser que se establezca un tamaño particular que represente el
tamaño menos ideal. Se deben realizar suficientes estudios para confirmar que
la distribución de calor es uniforme y reproducible en toda la cámara. Cada
estudio debe emplear un mínimo de 10 (diez) sensores de temperatura (calibrados
antes y después de su uso en el estudio) que serán expuestos en el medio
esterilizante dentro del autoclave.
Los estudios de distribución de calor
se deberán repetir siempre que exista cualquier alteración en la configuración
de la carga o cualquier modificación en el autoclave que pudiera alterar la
distribución de calor.
H-4.4.1.2. Colocación de los sensores de
temperatura en la carga.
Se debe distribuir un número adecuado
de sensores de temperatura en el espacio geométrico del autoclave para que las
zonas verticales y horizontales queden representadas. Colocar uno de los
sensores de temperatura en posición próxima al sensor de temperatura del
registrador del autoclave. Cada sensor debe ser ubicado en posición definida y
debe permanecer en esta posición durante todo el estudio asegurado por
dispositivos de fijación y no debe estar apoyado sobre los productos o las
superficies internas del autoclave.
H-4.4.1.3. Criterios de aceptación de los estudios de
distribución de calor.
a) No deben presentarse variaciones de temperatura
superiores a 2ºC por encima o debajo de la media de las temperaturas de todos
los sensores durante el período en que las cargas permanezcan a la temperatura
de la exposición.
b) Los sensores de temperatura se deben calibrar
antes y después de cada estudio y los resultados de las calibraciones no deben
presentar variaciones de temperatura de 0,5 ºC con respecto al termómetro de referencia.
c) Los parámetros operativos del ciclo de
esterilización se deben cumplir de acuerdo con las especificaciones del
protocolo.
d) La diferencia de temperatura entre el sensor del
registrador de temperatura del autoclave y el sensor en estudio no debe viajar
en más de 1,0ºC.
e) Debe haber por lo menos 9 (nueve) sensores
funcionando correctamente durante el estudio.
H-4.4.2. Estudios de penetración de calor
H-.4.4.2.1. Introducción.
Los estudios de penetración de calor e
realizan para asegurar que el envase más frío, dentro de una determinada
configuración de carga, esté expuesto consistentemente a suficiente letalidad
térmica. Deben ser realizados en cada cámara usando por lo menos las
configuraciones de carga máxima y mínima. Realizar el estudio en envases de diferentes
tipos y tamaños. El envase debe contener el volumen de llenado máximo con una
solución con características de calentamiento tan lentas como la de
calentamiento más lento de las soluciones a esterilizar.
H-4.4.2.2. Colocación de los sensores de temperatura
en la carga
a) En cada estudio se debe emplear un mínimo de 10
(diez) recipientes con un sensor de temperatura sumergido en la solución,
ubicados en sectores de la carga previamente determinados.
b) Para cada estudio pueden usarse 10 (diez) envases
inoculados con un indicador biológico, que pueden ser los mismos 10 (diez)
envases con sensores o unidades diferentes colocadas en posiciones adyacentes a
los sensores de temperatura.
c) Realizar un número suficiente de estudios de
penetración de calor para determinar si el valor de Fo preestablecido se
reproduce consistentemente a través de todo el autoclave.
Los datos empleados para calcular el
valor Fo deben ser obtenidos en aquella etapa del ciclo de esterilización donde
la temperatura en el interior del autoclave esté estabilizada y haya alcanzado
los valores especificados en el protocolo, de acuerdo con lo establecido en los
estudios de distribución de calor.
d) El sensor de temperatura se debe fijar al
recipiente lleno con solución, de modo que quede sumergido en el líquido sin
tocar las paredes del recipiente. Dado que cada recipiente posee
características propias, el fabricante debe desarrollar dispositivos
funcionales que garanticen que el sensor quede bien ubicado, además de
permanecer inmovilizado en la posición preestablecida dentro del autoclave,
durante los estudios. Las unidades con sensores no deben presentar pérdidas.
e) Distribuir las 10 (diez) unidades con sensores en
el espacio geométrico interior del autoclave, variando las posiciones de
estudio en estudio, de modo de ubicar posibles sectores de calentamiento lento.
Se recomienda el empleo de un programa de computación para calcular la posición
al azar de los sensores.
H-4.4.2.3. Criterios de aceptación de los
estudios de penetración del calor.
a) Los sensores de temperatura se deben calibrar
antes y después de cada estudio y los resultados de las calibraciones no deben
presentar variaciones de temperatura de 0,5 ºC con respecto al termómetro de referencia.
b) Se debe cumplir con los parámetros de operación del
ciclo de esterilización de acuerdo con lo especificado en el protocolo.
c) Deben funcionar correctamente por lo menos 9
(nueve) sensores durante el estudio.
d) Los recipientes con sensores de temperatura no
deben presentar pérdidas durante el estudio.
e) Los datos recolectados durante los estudios de
penetración de calor deben ser evaluados estadísticamente para determinar la
variación esperada en la letalidad generada por un determinado ciclo.
H-4.4.3. Cálculo del valor de F.
Existen varias fórmulas para el cálculo del valor
de F
a)
Utilizando la
fórmula
F = t. L
Donde: t = intervalo de tiempo entre las medidas de
temperatura
L = sumatoria de las tasas de
letalidad en cada intervalo.
Calcular las tasas de letalidad (L) a
intervalos de tiempo predeterminadas para cada sensor. Para mayor precisión se
recomienda que las lecturas de temperaturas sean realizadas a intervalos de 1
(un) minuto.
L = 10T1 T2/Z
Donde: L = Tasa de letalidad a cada intervalo de
tiempo.
T1 = temperatura del sensor en el instante T1.
T2 = temperatura de referencia 121ºC.
Z = Constante igual a 10ºC (valor de Z)
b) Una forma todavía más práctica de calcular el valor
de F es a través del uso de una Tabla de Tasas de Letalidad. Para cada lectura
de temperatura del sensor se busca el valor correspondiente de L en tabla. El
cálculo del valor de F se realiza sumando los valores de L encontrados en la
tabla, considerando Tasa de letalidad (L) para una temperatura de referencia de
121.11 ºC y un valor de Z = 10ºC.
L = minutos a 121,11 ºC por minuto a T ºC.
|
Temp ºC
|
0
|
0.1
|
0.2
|
0.3
|
0.4
|
0.50
|
0.6
|
0.7
|
0.8
|
0.9
|
|
90
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
|
91
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
|
92
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.001
|
0.002
|
|
93
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
|
94
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
0.002
|
|
95
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
|
96
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
0.003
|
0.004
|
0.004
|
0.004
|
0.004
|
|
97
|
0.004
|
0.004
|
0.004
|
0.004
|
0.004
|
0.004
|
0.004
|
0.005
|
0.005
|
0.005
|
|
98
|
0.005
|
0.005
|
0.005
|
0.005
|
0.005
|
0.005
|
0.006
|
0.006
|
0.006
|
0.006
|
|
99
|
0.006
|
0.006
|
0.006
|
0.007
|
0.007
|
0.007
|
0.007
|
0.007
|
0.007
|
0.008
|
|
100
|
0.008
|
0.008
|
0.008
|
0.008
|
0.008
|
0.009
|
0.009
|
0.009
|
0.009
|
0.010
|
|
101
|
0.010
|
0.010
|
0.010
|
0.010
|
0.011
|
0.011
|
0.011
|
0.011
|
0.012
|
0.012
|
|
102
|
0.012
|
0.013
|
0.013
|
0.013
|
0.013
|
0.014
|
0.014
|
0.014
|
0.015
|
0.015
|
|
103
|
0.015
|
0.016
|
0.016
|
0.017
|
0.017
|
0.017
|
0.018
|
0.018
|
0.019
|
0.019
|
|
104
|
0.019
|
0.020
|
0.020
|
0.021
|
0.021
|
0.022
|
0.022
|
0.023
|
0.023
|
0.023
|
|
105
|
0.024
|
0.025
|
0.026
|
0.026
|
0.027
|
0.027
|
0.028
|
0.029
|
0.029
|
0.030
|
|
106
|
0.031
|
0.032
|
0.032
|
0.033
|
0.034
|
0.035
|
0.035
|
0.036
|
0.037
|
0.038
|
|
107
|
0.039
|
0.040
|
0.041
|
0.042
|
0.043
|
0.044
|
0.045
|
0.046
|
0.047
|
0.048
|
|
108
|
0.049
|
0.050
|
0.051
|
0.052
|
0.054
|
0.055
|
0.056
|
0.057
|
0.059
|
0.060
|
|
109
|
0.062
|
0.063
|
0.064
|
0.066
|
0.067
|
0.069
|
0.071
|
0.072
|
0.074
|
0.076
|
|
110
|
0.077
|
0.079
|
0.081
|
0.083
|
0.085
|
0.087
|
0.089
|
0.091
|
0.093
|
0.095
|
|
111
|
0.097
|
0.100
|
0.102
|
0.104
|
0.017
|
0.109
|
0.112
|
0.115
|
0.117
|
0.120
|
|
112
|
0.123
|
0.126
|
0.128
|
0.131
|
0.135
|
0.138
|
0.141
|
0.144
|
0.148
|
0.151
|
|
113
|
0.154
|
0.158
|
0.162
|
0.166
|
0.169
|
0.173
|
0.177
|
0.182
|
0.186
|
0.190
|
|
114
|
0.194
|
0.199
|
0.204
|
0.208
|
0.213
|
0.218
|
0.223
|
0.229
|
0.234
|
0.239
|
|
115
|
0.245
|
0.251
|
0.256
|
0.262
|
0.268
|
0.275
|
0.281
|
0.288
|
0.294
|
0.301
|
|
116
|
0.308
|
0.315
|
0.323
|
0.330
|
0.338
|
0.346
|
0.354
|
0.362
|
0.371
|
0.379
|
|
117
|
0.388
|
0.397
|
0.406
|
0.416
|
0.425
|
0.435
|
0.446
|
0.456
|
0.467
|
0.477
|
|
118
|
0.489
|
0.500
|
0.512
|
0.523
|
0.536
|
0.548
|
0.561
|
0.574
|
0.587
|
0.601
|
|
119
|
0.615
|
0.629
|
0.644
|
0.659
|
0.674
|
0.690
|
0.706
|
0.723
|
0.739
|
0.757
|
|
120
|
0.774
|
0.792
|
0.811
|
0.830
|
0.849
|
0.869
|
0.889
|
0.910
|
0.931
|
0.953
|
|
121
|
0.975
|
0.997
|
1.021
|
1.044
|
1.069
|
1.094
|
1.119
|
1.145
|
1.172
|
1.199
|
|
122
|
1.227
|
1.256
|
1.285
|
1.315
|
1.346
|
1.377
|
1.409
|
1.442
|
1.475
|
1.510
|
|
123
|
1.545
|
1.581
|
1.618
|
1.655
|
1.694
|
1.733
|
1.774
|
1.815
|
1.857
|
1.901
|
|
124
|
1.945
|
1.990
|
2.037
|
2.084
|
2.133
|
2.182
|
2.233
|
2.285
|
2.338
|
2.393
|
|
125
|
2.448
|
2.505
|
2.564
|
2.624
|
2.685
|
2.747
|
2.811
|
2.877
|
2.944
|
3.012
|
|
126
|
3.082
|
3.154
|
3.228
|
3.303
|
3.380
|
3.459
|
3.539
|
3.622
|
3.706
|
3.792
|
|
127
|
3.881
|
3.971
|
4.063
|
4.158
|
4.255
|
4.354
|
4.455
|
4.559
|
4.665
|
4.774
|
|
128
|
4.885
|
4.999
|
5.116
|
5.235
|
5.357
|
5.481
|
5.609
|
5.740
|
5.873
|
6.010
|
|
129
|
6.150
|
6.293
|
6.440
|
6.590
|
6.744
|
6.901
|
7.061
|
7.226
|
7.394
|
7.566
|
|
130
|
7.743
|
7.923
|
8.108
|
8.296
|
8.490
|
8.687
|
8.890
|
9.097
|
9.309
|
9.526
|
|
131
|
9.747
|
9.974
|
10.207
|
10.445
|
10.688
|
10.937
|
11.192
|
11.452
|
11.719
|
11.992
|
|
132
|
12.271
|
12.557
|
12.850
|
13.149
|
13.455
|
13.769
|
14.089
|
14.418
|
14.753
|
15.097
|
|
133
|
15.449
|
15.808
|
16.177
|
16.554
|
16.939
|
17.334
|
17.737
|
18.151
|
18.573
|
19.006
|
|
134
|
19.449
|
19.902
|
20.365
|
20.840
|
21.325
|
21.822
|
22.330
|
22.850
|
23.382
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
23.927
H-4.5. Biovalidación por el método de
probabilidad de sobrevida
H-4.5.1. Introducción.
El método
establece los parámetros del ciclo en base al número de microorganismos
presentes en el producto antes de la esterilización (Biocarga) y a la
resistencia térmica de dichos microorganismos. La combinación del número y de
la resistencia térmica determinará la cantidad de calor requerida para alcanzar
la probabilidad de sobrevida microbiana en el producto a un valor no superior a
10-6 microorganismos. Generalmente este método se emplea en la
validación de ciclos de esterilización de productos termolábiles.
H-4.5.2.
Los estudios microbiológicos para la biovalidación deben incluir:
a) Programa de monitoreo microbiológico
ambiental.
b) Programa de monitoreo de la biocarga
pre esterilización y de determinación de la resistencia térmica de los
microorganismos.
c) Estudios de laboratorio de los
valores de D y Z de los bioindicadores.
d) Evaluación de los datos
microbiológicos con el objeto de asegurar que la letalidad del proceso responde
a las especificaciones proyectadas para la esterilización.
H-4.5.3. Metodología
H-4.5.3.1. Preparación de los envases inoculados
con bioindicador en la carga de esterilización
a) Usar 10 (diez) envases llenos con la
solución recientemente preparada, e inocular cada envase con la suspensión de
esporos en magnitud cercana a 106 a 107.
Los
microorganismos a utilizar como bioindicadores deben ser seleccionados a partir
de la biocarga de preesterilización en base a su resistencia al calor.
b) Los sensores de temperatura deben ser
colocados en los envases inoculados o bien en posiciones adyacentes a los
mismos.
H-4.5.3.2. Preparación y recuento de los
controles positivos
a) Preparar 2 (dos) envases inoculados
de modo similar a lo indicado en 4. 5. 3. 1. (A) para control positivo.
b) Homogeneizar la solución dentro de los
envases agitándola vigorosamente por 3 (tres) a 5 (cinco) minutos.
c) Pipetear 1.0 mL de muestra y efectuar
una serie de diluciones 1:10 en solución estéril de cloruro de sodio al 0.9%,
hasta obtener una concentración final de aproximadamente 30-300 esporos por mL.
Inocular las diluciones por triplicado, en cajas de Petri con medio de cultivo
e incubar por 24 (veinticuatro) horas - 72 (setenta y dos) horas.
d) Efectuar el conteo de unidades formadoras
de colonias (UFC).
NOTA: Los medios de cultivo a ser
utilizados y las condiciones del ensayo e incubación dependerán del tipo de
microorganismo utilizado como bioindicador.
H-4.5.3.3.
Ensayo de los envases inoculados
a) Recoger los envases inoculados y
esterilizados inmediatamente después de completado el ciclo de esterilización.
b) Considerar los envases como muestras para ensayo de
esterilidad, efectuando el ensayo por el método de filtración por membrana, en
un ambiente adecuado.
c) Incubar durante 7 (siete) días la membrana en el
medio de cultivo y condiciones indicadas para el microorganismo utilizado como
bioindicador.
d) Efectuar el recuento de UFC o utilizar el método
de fracciones negativas a fin de determinar el número de sobrevivientes.
H-4.5.3.4. Evaluación de los resultados
Determinar la letalidad efectiva del
ciclo de esterilización mediante la ecuación siguiente:
Fzt = Dzt'
(log10A - log10B)
Donde:
Dzt = valor
determinado previamente por estudios de laboratorio para el bioindicador
empleado.
A = número de esporos inoculados por envase
B = número de microorganismos sobrevivientes por
envase
Cuando se utilice el método de fracción negativa:
B
= 2.303 log10 (n/q)
Donde:
n
= número total de recipientes inoculados
q
= número de recipientes inoculados negativos
H-4.6.
Calibración de los indicadores biológicos para el método de sobremuerte
H-4.6.1. Introducción.
Los indicadores biológicos son
utilizados en la validación para medir la letalidad producida por el ciclo de
esterilización, de manera de asegurar que la probabilidad de sobrevida
microbiana sea no superior a 1 x 10-6. Los indicadores biológicos
que se utilicen deben ser calibrados antes de su uso, sean ellos de origen
comercial o preparados en el laboratorio.
Cuando la letalidad del ciclo sea
suficiente para producir 12 log de reducción en microorganismos que tengan un
valor D de 1 (un) minuto, no será necesario efectuar estudios rutinarios de
biocarga ni de resistencia térmica en productos aislados.
H-4.6.2. Microorganismos utilizados como
indicadores biológicos.
Debido a su elevada resistencia al
calor, se utilizan frecuentemente como microorganismos de prueba tanto el Clostridium
sporogenes como el Bacilus stearothermophilus.
El número y resistencia térmica de
poblaciones de esporos son criterios importantes en la selección de indicadores
biológicos para el proceso de esterilización por vapor.
H-4.6.3. Tipos de soporte.
El tipo de soporte es un factor
importante en la determinación de los valores D y Z de los indicadores
biológicos debido a que puede afectar la resistencia de los mismos.
Frecuentemente se utilizan los siguientes tipos de
soporte:
a) microorganismos suspendidos en la solución
b) tiras de papel
c) material de la misma composición que el producto a ser esterilizado
Los valores de D y Z deben ser
determinados utilizando el mismo tipo de soporte usado para controlar el ciclo
de esterilización.
H-4.6.4. Determinación del valor D (reducción
decimal del indicador biológico)
H-4.6.4.1. Metodología:
a) Inocular muestras de SPGV, por duplicado, con una
suspensión que contenga un número conocido de esporos del indicador biológico
seleccionado.
b) Preparar controles positivos para determinar el
número de esporos en la suspensión (o tira, etc) a ser ensayada. El número de
esporos necesarios deberá basarse en el esquema de cada experiencia, pero
generalmente es del orden de 104 a 107 esporos por
muestra.
c) Exponer por lo menos 5 (cinco) muestras a una
temperatura determinada por, por lo menos, 3 (tres) intervalos de tiempo
diferentes, a fin de determinar el número de sobrevivientes (cálculo del valor
D).
H-4.6.4.2. Cálculo.
El valor D, o tiempo de reducción
decimal, a determinada temperatura, es el tiempo necesario para inactivar el 90
% de la población microbiana del producto por el método de la fracción negativa
de acuerdo a lo descripto en 4. 3. 3. 2 (a) y (b) de esta norma.
H-4.6.5. Determinación del valor de Z (número de grados
de temperatura necesarios para obtener la reducción logarítmica del valor D).
Se define el valor de Z como el número
de grados de temperatura necesarios para cambiar el valor D por un factor de
10. Se puede determinar mediante lo establecido en el punto 4.3.4. de esta
norma.
H-4.6.6. Determinación del valor F0
Se recomienda el uso del Bacilus
stearothermophilus como indicador biológico para ciclos de esterilización
de productos resistentes al calor, debido a su elevada resistencia térmica. El
valor D de esto indicadores biológicos debe ser mayor de 1 (un) minuto.
F0 = D121 (log10 A
- log B)
Donde:
F0 =Tiempo a 121ºC necesario para reducir en un 90% la población de microorganismos.
A = Concentración inicial de esporas por
recipientes.
B = Nivel máximo aceptable para una
probabilidad de sobrevida de 1 x 10-6.
H-4.7. Biovalidación por el método de sobremuerte
("overkill")
H-4.7.1. Introducción.
El método establece los parámetros del
ciclo en base a la resistencia térmica de los indicadores biológicos utilizados
en el proceso de validación y la concentración de esporos presentes en el
soporte utilizado en el ensayo.
Debido a su elevada resistencia
térmica se utilizan frecuentemente como indicadores biológicos el Cl,
sporogenes y el B, stearothermophilus. También se pueden utilizar otras
especies bacterianas formadoras de esporos, siempre que sean debidamente
calibradas.
H-4.7.2. Los estudios microbiológicos
para biovalidación deben incluir:
a) Soporte inoculado con un volumen conocido de una
suspensión calibrada de esporo
b) Controles positivos para verificar el conteo inicial.
c) Estudios microbiológicos a efectuar durante los
ensayos de penetración del calor en 10 (diez) puntos del autoclave, de manera
de asegurar que se hallen representados los puntos fríos del autoclave.
d) Carga patrón idéntica a la especificación para el
uso rutinario de producción.
H-4.7.3.
Metodología
H-4.7.3.1.
Ensayo del
producto con indicadores biológicos:
a) Inocular con el indicador biológico 10 (diez)
envases que contengan el producto.
b) Retirar los recipientes inoculados inmediatamente
después de la esterilización.
c) Considerar los recipientes como muestras para
ensayo de esterilidad y realizar los ensayos en un ambiente adecuado.
d) Inocular en un medio de cultivo apropiado para el
crecimiento de microorganismos (Typticase Soy Broth), colocar a la temperatura
ideal para su crecimiento y dejar por un período no inferior a las 72 (setenta
y dos) horas.
e) Registrar el número de indicadores biológicos con
resultados positivos y negativos en cuanto a desarrollo de microorganismos.
H-4.7.3.2. Evaluación de los resultados.
Calcular la letalidad efectiva del
ciclo de esterilización mediante la ecuación:
Fzt= (log10 A -
log10 B)
Donde:
Dzt = Valor previamente
determinado por estudios de laboratorio para el indicador biológico utilizado.
A = número de esporos inoculados por recipiente
B = número de microorganismos sobrevivientes por
envase
Cuando se utilice el método de la
fracción negativa debe usarse la siguiente ecuación:
B
= 2.303 log10 (n/q)
Donde:
n = número total de recipientes inoculados
q = número de recipientes inoculados
negativos en cuanto al crecimiento de microorganismos
H-4.8. Criterios de aceptación de la biovalidación
H-4.8.1. Se considera aceptable el estudio de
penetración del calor cuando, además de los requisitos establecidos en 4.4.2.3,
todas las unidades inoculadas sometidas al ensayo de esterilidad no mostraron
crecimiento de microorganismos.
H-4.8.2. Si ocurriera la presencia de un falso
positivo se podría aceptar la biovalidación si se demostrara, luego del ensayo
microbiológico, que el microorganismo positivo es diferente del utilizado como
indicador biológico.
H-4.8.3. El recuento de microorganismos de los recipientes
de control debe ser igual o mayor de 4 x 105.
H-4.8.4. Por lo menos 9 (nueve) unidades de las inoculadas
deben estar íntegras (sin pérdida de solución).
H-4.9. Condiciones de carga del autoclave.
Los estudios de calificación deben
hacerse utilizando la carga máxima y mínima del autoclave. Si los resultados
obtenidos mostraran variaciones significativas en la distribución del calor,
también deberán efectuarse estudios con cargas intermedias.
H-4.10. Modificaciones en el producto y/o su envase
H-4.10.1. Se requerirá una nueva validación si
hubiera cambios significativos en el recipiente
H-4.10.2. También se requerirán nuevos estudios
de validación que confirmen la eficacia del proceso en el caso de que hubiera
cambios en la composición, viscosidad, volumen de la solución o de la capacidad
del recipiente que pudieran afectar los valores de transferencia del calor.
H-4.11. Evaluación de los resultados.
La documentación, incluyendo
protocolos, datos obtenidos de los instrumentos, procedimiento, validación y
calificación, deben ser revisados por los responsables de garantía de calidad
que aprobarán por medio de una certificación formal la validación del proceso
para esterilizar con seguridad los productos especificados en el protocolo. En
caso de que los datos no sean considerados lo suficientemente confiables, se
solicitarán nuevos estudios antes de la liberación del proceso en la etapa de
validación.
H-5. CRITERIOS DE VALIDACION
H-5.1.
Se debe planear un
estudio sencillo de calificación periódica con el objeto de detectar cambios
inadvertidos. El período de tiempo debe estar determinado por las características
del proceso de producción.
H-5.2. En los intervalos de tiempo que se especifiquen se
deben recalificar equipamientos y procedimientos, empleando métodos
ingenieriles o microbiológicos.
H-5.3. Después de efectuar cualquier modificación en el
equipo debe efectuarse un estudio sencillo de calificación a fin de demostrar
que el proceso de esterilización no fue alterado.
H-5.3.1. El regular mantenimiento preventivo o correctivo o
la reposición de partes equivalentes no obligan a nuevos estudios de revalidación.
H-5.3.2. Si el mantenimiento implicara
componentes electrónicos o de control, deberá efectuarse una revalidación a
menos que se pueda demostrar que no se producirán alteraciones significativas
con motivo de dicha acción.
H-5.4. De no efectuarse una revalidación por alguna de
las razones expuestas, se deberá realizar la misma con intervalos no superiores
a un año.
H-6.
RECERTIFICACION
Deberá
efectuarse luego de cada estudio de revalidación, con el objeto de revisar,
documentar y aprobar los estudios efectuados.
ANEXO I
TAPONES DE ELASTOMERO PARA SOLUCIONES
PARENTERALES DE GRAN VOLUMEN
I.1
OBJETIVO
I.2
DEFINICIONES
I.3
CONDICIONES GENERALES
I.4
CONDICIONES ESPECÍFICAS
I.5
CRITERIOS PARA LA REVALIDACIÓN
I-1
OBJETIVO
Esta norma fija las condiciones exigibles relativas a los
aspectos físicos, químicos y biológicos para elastómeros naturales o
sintéticos, utilizados como tapones de recipientes conteniendo SPGV.
I-2. CONDICIONES GENERALES
Un tapón de
elastómero natural o sintético para recipientes con SPGV debe:
a) Estar sujeto a exigencias especiales,
especialmente en lo referente a componentes solubles, debido a su contacto con
medicamentos de las más diversas composiciones.
b) Ser analizado por el fabricante en cuanto a la
posibilidad de cesión de sus componentes y a la incompatibilidad con el
producto.
c) Ser usado una única vez.
d) Tener la garantía del fabricante de que todas las
piezas de una entrega han sido fabricadas con material de la misma formulación
y que presentan las mismas propiedades. Cada lote debe ser examinado por
separado.
e) Estar libre de materias extrañas, polvo, fibras,
partículas de elastómero, pigmentos y de cualquier sustancia que pudiera ceder
sustancias tóxicas, pirogénicas, de acción bacteriostática o bactericida y hemolíticas.
f) Ser almacenado a temperatura entre 0 ºC y 30 ºC al abrigo de la luz y de la radiación ultravioleta.
g) Ser lavado, antes del uso, de acuerdo con las
instrucciones específicas provistas por el fabricante y que dependen del
material con el cual fue fabricado.
I-3.
CONDICIONES ESPECIFICAS
I-3.1 Requisitos físicos.
I-3.1.1. Color.
El color de cada lote de tapones de elastómero debe
ser uniforme.
I-3.1.2. Dureza.
La dureza "Shore", de
acuerdo con el método descripto en el ítem I-4. 3. 2, no debe variar de 15% de
la del patrón, dentro del período de aplicación, garantizado por el fabricante,
y sin la influencia de otras sustancias.
I-3.1.3. Fragmentación.
Sólo para tapones que poseen un sitio
para ser perforado con agujas hipodérmicas. Debe presentar, como máximo, una
media de 5 fragmentos por tapón de elastómero, de acuerdo con el método
descripto en I-4. 3. 3.
NOTA: La aparición de fragmentos depende de numerosas
particularidades, inclusive del modo de punción. Aún utilizando una aguja hipodérmica
nueva, con buena punta y bien pulida, pueden producirse fragmentos.
I-3.1.4. Penetrabilidad.
Sólo para tapones que poseen un sitio
para perforar con agujas hipodérmicas. La fuerza necesaria para lograr la
penetración de una aguja hipodérmica en tapones de elastómero no debe ser
superior a 1000g, de acuerdo con el método descripto en I-4.3.4.
I-3.1.5. Compatibilidad con productos inyectables.
Realizar el ensayo sólo con cada
producto nuevo según lo indicado en I-4.3.5.
I-3.1.6. Empaque e identificación.
Los tapones deben ser empacados
limpios, protegidos del polvo y de la luz, identificados con un rótulo que
indique:
a) Declaración de contenido
b) Fecha de fabricación
c) Número de lote
d) Identificación del fabricante
I-3.2.
Requisitos químicos.
Los tapones
de elastómero utilizados en los recipientes de SPGV deben cumplir las
exigencias de la Tabla I.
TABLA I
|
ANALISIS
|
ESPECIFICACIONES
|
|
Aspecto de
la solución extractiva
|
Pasa el
ensayo
|
|
Metales
pesados
|
Más. 2.0 ppm
|
|
Amoino
|
Más. 2.0
ppm
|
|
Cloruros
|
Máx. 4.0 ppm
|
|
Sulfuros
volátiles
|
Máx. 0.154 mg
Na2S/20 cm2
|
|
Zinc soluble
|
Máx. 5.0 ppm
|
|
Sustancias
reductoras
|
Máx. 1.5 mL de
Na2S2O3 0.01 N / 10 mL
|
|
Acidez o
alcalinidad
|
Máx. 0.8 mL Hcl
0.01N ó 0.3mL NaOH 0.01 N/ 20 mL
|
|
Residuo seco
|
Máx. 4 mg /
100 mL de extractivo
|
|
Absorbancia
|
Máx. 0.2
entre 220 y 360 mm
|
I-3.3 Requisitos biológicos
a) Sustancias piretógenas: los tapones de
elastómeros no deben liberar a las soluciones sustancias capaces de ejercer
efectos piretogénicos.
Requisito para el ensayo de piretógenos: negativo.
Límite para el ensayo de endotoxinas bacterianas:
menor a 0.5 UE/ml.
b) Toxicidad: los tapones de elastómero no
deben liberar a las soluciones sustancias capaces de ejercer efectos tóxicos.
c) Acción bacteriostática o bactericida: Los tapones de elastómero no deben
tener acción bacteriostática o bactericida.
I-4.
METODOS DE ANALISIS
I-4.1. Periodicidad.
Los ensayos deben ser realizados sobre cada lote, a
menos que exista un programa formal de certificación de proveedores en el cual
la periodicidad esté definida.
I-4.2. Muestreo.
Los tapones de elastómero deben ser
muestreos lote a lote según un método estadístico Ön
+ 1, dónde n es igual al número de envases y en cantidad suficiente
para, como mínimo, poder realizar dos repeticiones de ensayos físicos, químicos
y biológicos.
I-4.3.
Ensayos físicos.
Las muestras
destinadas a ensayos físicos deben ser lavadas dos veces con agua deionizada,
esterilizadas durante 30 minutos en vapor de agua saturado a presión a 121 ºC +/- 2 ºC, colocados en estufa a 60 ºC como máximo durante 60 minutos y guardados en frasco de
vidrio cerrados hasta el inicio de los ensayos.
I-4.3.1. Color.
Examinar las muestras luego de su
enfriamiento, observando la presencia de manchas o decoloración.
I.4.3.2. Dureza.
Examinar la dureza del elastómero,
realizando las lecturas con un Durómetro en partes planas, o secciones
especiales del tapón. En caso de ser necesario la lectura debe hacerse sobre
una superficie plana con un espesor de 6,25 mm obtenida por superposición de una cantidad suficiente de trozos planos cortados. El durómetro debe ser calibrado
con frecuencia con un bloque patrón provisto con cada instrumento.
I-4.3.3. Fragmentación.
a) Llenar hasta a mitad con agua libre de partículas,
20 frascos y taparlos de forma apropiada con tapones en ensayo.
b) Usando una aguja hipodérmica 21 G (0,813 mm x 38 mm) y de punta normal, perforar 5 veces una cara del tapón, con una velocidad de
20 +/- 1 cm/min asegurándose que las 5 perforaciones estén dentro de un círculo
de 5 mm de diámetro y tan equidistantes unas de otras como sea posible.
c) Luego de la 5a perforación, sin retirar
la aguja del tapón, conectarle a la aguja una jeringa hipodérmica limpia,
conteniendo cerca de 1 mL de agua libre de partículas e inyectar el agua en el
frasco.
d) Retirar la aguja y examinar cuidadosamente para
verificar si la punta del bisel quedó despuntada. Si esto fuera evidente,
descartar el frasco y su contenido y sustituirlo por uno nuevo.
e) Repetir el ensayo sobre los otros frascos tapados,
usando una aguja nueva para cada tapa.
f) Filtrar el agua de cada frasco, a través de un
filtro Buchner usando papel de filtro de color constante (si el tapón en ensayo
fuera blanco el papel de filtro debería ser teñido con un color constante, por
ejemplo con azul de metileno).
g) Contar el número de fragmentos sin ayuda
artificial (como por ejemplo lentes de aumento).
I-4.3.4. Penetrabilidad.
Tapar 5 frascos con los tapones a ser
ensayados. Penetrar, cada uno de ellos con una aguja hipodérmica 21 G (0.813 mm x 38 mm) a una velocidad de 20 +/- 1 cm/min, usando un dispositivo para medir el
esfuerzo de penetración con una precisión de +/-25 g para una factura de 100 g.
Asegurar que la penetración de la
aguja sea en la posición perpendicular a la superficie del tapón. Registrar la
fuerza máxima para cada penetración.
I-4.3.5. Compatibilidad con productos inyectables (Sólo
para productos nuevos o formulaciones nuevas del tapón)
a) Para la preparación de las muestras de ensayo
emplear como mínimo 32 y como máximo 50 tapones preparados de acuerdo a lo
indicado en I-4. 3, para cada producto inyectable con el cual se pretende
ensayar su compatibilidad y usar frascos del mismo tipo del que normalmente se
emplean con el producto.
b) Preparar el mismo número de frascos control con
tapones lavados, autoclavados y previamente aprobados.
c) Llenar los frascos con la solución
con la cual van a ser usados, en las condiciones normales de envasado del
producto.
d) Cerrar los frascos adecuadamente y examinar para
verificar si presentan cualquier alteración (por ejemplo color, turbidez,
etc.). Si el examen no fuera satisfactorio, descartarlos y preparar nuevas
muestras.
e) Esterilizar los frascos en las condiciones
recomendadas para el producto, colocando la mitad de ellos en posición
invertida.
f) Mantener los frascos con la mitad en posición
invertida, en las condiciones de almacenamiento indicadas e inspeccionar
visualmente a cada uno de los períodos recomendados en la tabla siguiente, para
observar la presencia de cualquier alteración visible.
|
Condiciones de almacenamiento
|
Nº de muestras de ensayo y control
|
Inspección (*) (meses)
|
|
4 ºC (heladera)
|
4+4
|
1, 3, 6, 9 y 12
|
|
25 ºC
|
8+8
|
1, 3, 6, 9 y 12
|
|
38 ºC
|
8+8
|
1, 3, 6, 9 y 12
|
|
38 ºC/16 hs
y 4 ºC/8 hs alternadamente, a
90 % - 100
% de HR
|
8+8
|
1, 3, 6, 9 y 12
1, 3, 6, 9 y 12
|
|
50 ºC u otra temperatura conveniente
|
4+4
|
1, 2 y 3
|
(*) Inspección en los intervalos luego del
inicio del ensayo.
Observaciones:
1. El almacenamiento a 25 1/2C y 38 1/2C es el de
mayor importancia para la evaluación de la compatibilidad de los tapones con
los productos.
2. El almacenamiento a 50 ºC o temperaturas más altas tiene importancia sólo en los casos en que la incompatibilidad sólo
pueda ser evidenciada a temperaturas por encima de 38 1/2C y puede ser usado
como un ensayo acelerado.
3. El almacenamiento a 4 °C no es importante para la evaluación de la compatibilidad, pero es útil para evaluar si la
apariencia inicial sufre alguna alteración por acción de la temperatura.
4. El almacenamiento con alto tenor de humedad
(90%-100% de HR) está incluido por el posible efecto adverso en la
penetrabilidad y fragmentación de los tapones en ensayo.
g) Evaluar la solución de producto ensayado para
verificar si continúa cumpliendo sus especificaciones (potencia, toxicidad,
contenido bacteriostático, etc.), al mismo nivel que los frascos control.
h) Inspeccionar los tapones observando decoloración,
porosidad, hinchamiento o cualquier otra señal de deterioro.
Los tapones deben ser ensayados
también en cuanto a penetrabilidad y fragmentación. Si los tapones se
presentaran decolorados luego del contacto con el líquido, deben ser
reensayados luego de una noche de reposo para su secado.
La ocurrencia de cualquiera de las
citadas anormalidades debe ser considerada como evidencia de incompatibilidad
entre el tapón y el producto.
I.4.3.6. Empaque e identificación:
El empaque e identificación de los
tapones en el momento de su recepción, debe cumplir con los requisitos
indicados en I-3.1.6.
I-4.4. Ensayos químicos:
Los ensayos químicos se llevarán a
cabo de acuerdo con lo establecido en la edición vigente de la Farmacopea Europea.
I-4.5. Ensayos biológicos
I-4.5.1. Sustancias piretógenas. Utilizar
la metodología descripta en el Anexo L "Ensayos biológicos", L-2
Ensayo de piretógenos y L-3 Ensayo de endotoxinas bacterianas.
Se puede realizar cualquiera de los dos ensayos.
I-4.5.1.1. Preparación de la muestra para el
ensayo de piretógenos
a) Usar una cantidad de muestra correspondiente a una
superficie total de 100 cm2+/-5cm2 para preparar 200 mL
de solución extractiva.
b) Colocar las muestras en un frasco previamente
lavado con agua para inyectables, estéril y apirogénica. Adicionar 200 mL de
solución fisiológica de cloruro de sodio y tapar el frasco adecuadamente.
Extraer durante 30 minutos a 121 ºC +/- 2 ºC.
c) Enfriar a temperatura ambiente y llevar al volumen
originalmente contenido en el frasco, con agua para inyectables estéril y
apirogénica.
I-4.5.2. Ensayos de toxicidad.
Los tapones de elastómero deben ser
sometidos a los ensayos de reactividad biológica, de acuerdo con lo indicado en
Anexo L-5: Ensayos de reactividad biológica.
I-4.5.2.1. Preparación de muestras para el ensayo de
reactividad biológica in vitro
a) Usar una cantidad de muestra correspondiente a una
superficie total de 100 cm2+/-5 cm2 para preparar 200 mL
de solución extractiva.
b) Colocar las muestras en un frasco y lavar dos veces
con agua para inyectables estéril y apirogénica a 60 ºC dejando escurrir las aguas de lavado. Adicionar 200 mL de solución fisiológica de cloruro de
sodio. Tapar los frascos con papel de aluminio y extraer durante 30 minutos a
121 ºC+/-2 ºC.
c) Enfriar a temperatura ambiente y llevar el volumen
originalmente contenido en el frasco, con agua para inyectables estéril y
apirogénica.
I-4.5.3. Ensayo para verificar la actividad
bacteriostática o bactericida
I-4.5.3.1. Preparación de la muestra
a) Usar una cantidad de muestra correspondiente a una
superficie total de 100 cm2 para preparar 200 mL de solución extractiva.
b) Colocar las muestras, previamente
lavadas (dos veces) con agua para inyectables estéril y apirogénica a 560 ºC, en un frasco, previamente esterilizado, con 200 mL de una solución fisiológica de cloruro de
sodio, estéril y apirogénica. Rápidamente extraer, durante 30 minutos, a 121 ºC +/-2 ºC.
Observación: El líquido debe ser
incoloro y no debe presentar turbidez.
I-4.5.3.2. Procedimiento para el ensayo
a) Mezclar 5 mL de la muestra (solución de ensayo) con
5 mL de caldo de carne (caldo nutriente) de concentración doble en un tubo de
ensayo previamente esterilizado.
b) Preparar una solución control, mezclando 5 mL de
caldo de carne, con 5 mL de agua para inyectables estéril en un tubo de ensayo
previamente esterilizado.
c) Inocular ambos tubos de ensayo con 0.2 mL de un
cultivo de Microccocus pirogenes var.aureus de una antigüedad de 16 horas,
diluido en proporción 10: 10.000.
Incubar a 37 ºC durante 72 horas.
d) Luego de la incubación, el contenido de los 2 tubos
de ensayo debe presentar un evidente crecimiento de las bacterias de ensayo.
e) Comprobar el crecimiento de las bacterias de ensayo
mediante la realización de un subcultivo en el medio de un cultivo apropiado.
Interpretación: El crecimiento de gérmenes indica
ausencia de actividad bacteriostática o bactericida en la muestra.
I-5.
ACEPTACION O RECHAZO
Los tapones de elastómero serán aceptados siempre
que cumplan las exigencias de esta Norma, en caso contrario, serán rechazados.
ANEXO J
ESTABILIDAD
J.1 OBJETIVO
J.2 DEFINICIONES
J.3 MÉTODOS
J.4 DETERMINACIÓN DEL PERÍODO DE
VIDA ÚTIL POR MÉTODO DE ESTABILIDAD ACELERADA
J.5 CONSIDERACIONES
GENERALES
J-1. OBJETIVO
Esta norma establece lineamientos generales para
los estudios de estabilidad y criterios para la determinación del período de
vida útil (fecha de vencimiento) de las SPGV.
J-2.
DEFINICIONES
A los fines
de esta norma se adoptan las siguientes definiciones:
J-2.1. Estabilidad:
Es la capacidad de un producto de
mantener sus características originales conforme a sus especificaciones de
pureza, calidad y potencia.
El estudio de
la estabilidad se realiza en una fase previa a la comercialización de un
producto nuevo o cuando se efectuaran cambios en el proceso de elaboración.
J-2.1.1. Características del producto:
a) Características físicas: deben conservarse las propiedades
físicas tales como: aspecto, limpidez, ausencia de partículas extrañas, pH, y
hermeticidad.
b) Características químicas: la degradación de sus principios activos
no debe superar el 10% y no deben presentarse sustancias extrañas en la
composición del producto.
b) Características biológicas: el medicamento debe mantenerse
estéril, libre de pirógenos y atóxico.
J-2.2. Período de vida útil:
Es el tiempo
(en días, meses y años) durante el cual un medicamento se mantiene, en la
totalidad de su período de almacenamiento y de uso, dentro de los límites
especificados y con las mismas propiedades y características que poseía en el
momento de la fabricación.
J-3. METODOS
J-3.1.
Estabilidad natural:
Consiste en mantener un producto a una
temperatura de 25 ºC +/- 1 ºC, en condiciones de humedad y exposición a la luz
conocidas controlando las características físicas, químicas y biológicas
inicialmente y a intervalos regulares durante un período de tiempo determinado.
A través de este estudio se determinará el período de vida útil presupuesto,
que estará limitado al tiempo durante el cual se mantengan las características
físicas, químicas y biológicas del producto, encontrándose la concentración de
los principios activos dentro de las especificaciones de la monografía.
J-3.2. Estabilidad acelerada:
Se utiliza
este método para estimar el período de vida útil hasta tanto se completen los
estudios de estabilidad natural.
Consiste en
mantener grupos de muestras de SPGV a temperaturas elevadas, controlando las
características físicas, químicas y biológicas inicialmente y conforme a la
periodicidad que se sugiere en la siguiente tabla.
|
Temperatura ºC
|
Período (días)
|
|
40
|
30
|
|
60
|
|
90
|
|
50
|
10
|
|
20
|
|
30
|
|
60
|
3
|
|
7
|
|
10
|
J-4. DETERMINACION DEL PERIODO DE VIDA UTIL POR EL METODO DE ESTABIIDAD
ACELERADA
El período de vida útil se establece inicialmente por
medio de los resultados obtenidos en el estudio de estabilidad acelerada,
teniéndose en cuenta la velocidad de reacción (descomposición) y el orden de
reacción.
J-4.1. Determinación del orden de la reacción por el
método gráfico.
Representar
gráficamente los valores experimentales obtenidos de las concentraciones en función
del tiempo requerido para alcanzarlas.
Cuando se
obtiene una recta al graficar concentraciones versus tiempo, la reacción es de
orden 0. Si para obtener una recta es necesario representar el logaritmo de la
concentración remanente en función del tiempo, la reacción es de primer orden,
y si es necesario representar la inversa de la concentración en función del
tiempo para obtener una relación lineal, la reacción es de segundo orden.
J-4.2. Determinación de la velocidad de reacción
(descomposición).
La ecuación
de Arrhenius muestra la influencia de la temperatura sobre la velocidad de
reacción (descomposición).
Log K=log A - DE . 1
2,303 . R T
Donde:
K: Constante
de la velocidad de reacción (descomposición)
A: Factor de frecuencia
DE: Energía de activación
R: Constante de los gases perfectos (1,987 cal. mol-1
. ºK-1)
T: Temperatura absoluta (ºK)
La constante dependerá del orden de reacción.
Para la reacción de orden cero la constante es =
log A + 0,30103 - log Co
Para la reacción de primer orden la constante es =
log A - log 0,693
Para la reacción de segundo orden la constante es =
log a + log A
Donde Co = la mitad de la concentración inicial
a: concentración inicial
Cálculo de K por el método gráfico.
Construir un gráfico representando en la ordenada
los valores de log K y en la abcisa 1/T . 103 obteniéndose por
simple extrapolación el valor de log K a 25 ºC.
J-4.3. Cálculo para determinación del período de
vida útil (tm)
Considerando una reacción de orden cero
Tm= Xo – X
K
Donde Xo: Concentración inicial de producto.
X: Concentración final aceptable (90%)
K: Constante de velocidad de reacción
(descomposición) a 25 ºC +/- 1 ºC.
Considerando una reacción de primer orden
Tm= 2,303 . log Xo
K K
Considerando una reacción de segundo orden
Tm= Xo – X
Xo . X . K
Con la
aplicación de una de las fórmulas arriba citadas, se obtiene el período de vida
útil expresado en días.
J-5
CONSIDERACIONES GENERALES
1) Los métodos de valoración deben ser
suficientemente específicos y sensibles como para poder detectar una eventual
degradación de los principios activos.
2) Se deberá utilizar un número
adecuado de lotes y muestras para permitir la obtención de datos
estadísticamente válidos.
3) Se deberá verificar que a lo largo del
período de vida útil propuesto no aparezcan sustancias originadas en el envase
en cantidades tales que afecten las características físicas, químicas y
biológicas del producto.
4) Se debe tener en cuenta que los
métodos de cálculo para determinar el período de vida útil solo consideran las
características químicas del principio activo.
ANEXO L
ENSAYOS BIOLOGICOS
L-1 OBJETIVO
L-.2 ENSAYO
DE PIRETÓGENOS
L-.3 ENSAYO
DE ENDOTOXINAS BACTERIANAS
L-4 ENSAYO
DE ESTERILIDAD
L-.5 ENSAYOS
DE REACTIVIDAD BIOLÓGICA
L-5.1 IN VITRO
L-5.2 IN VITRO
L-6 ENSAYO
DE CONTAMINACIÓN MICROBIANA
L-1. OBJETIVO
Esta norma establece los procedimientos para la
realización de los ensayos biológicos aplicables al control de las SPGV.
L-2. ENSAYO DE PIRETOGENOS
L-2.1. Condiciones generales.
El ensayo de piretógenos se fundamenta
en la medida de la variación de la temperatura corporal de los conejos que son
inoculados, por vía endovenosa, con una solución estéril de la sustancia en
análisis.
Para los
productos que requieran una preparación preliminar o que necesiten condiciones
especiales de administración, seguir las recomendaciones de las monografías
específicas.
L-2.2. Materiales y disolventes.
Utilizar jeringas, agujas y material
de vidrio apirogénico (tratados a 250 ºC por un tiempo mínimo de 30 (treinta) minutos a 200 ºC durante un mínimo de 1 (una) hora). Preparar todos los
disolventes y soluciones de lavado de materiales con agua estéril y
apirogénica.
L-2.3. Control, selección y preparación de los
animales
a) Organizar una ficha propia para cada animal, registrando:
-Edad
-Sexo
-Peso encontrado semanalmente y en el día anterior
a su utilización para el ensayo;
-Fecha en que el animal fue sometido al último
ensayo y resultado del mismo.
b) Utilizar animales con un peso corporal no inferior
a 1,5 Kg., con venas auriculares buenas y poco ramificadas, alimentados con
ración exenta de antibióticos y que no pierdan peso dentro de la semana. Los
animales que nunca hayan sido utilizados para ésta prueba deberán someterse a
un período de acostumbramiento y control previo. Para ello se colocarán 2 hs.
diarias en el cepo durante una semana. Durante la semana siguiente se
registrará su temperatura a los 60 y 120 minutos de colocados en el cepo.
Finalmente se realizará una prueba con Solución Fisiológica apirogénica; no
debiendo observarse aumentos de temperatura superiores a 0.6 ºC.
c) No utilizar animales para el ensayo de piretógenos
con un intervalo menor a 3 días; en caso de que el animal haya presentado una
reacción pirogénica, no volver a utilizarlo antes de por lo menos 14 (catorce)
días.
d) Los conejos destinados al ensayo de piretógenos que
no hubieran perdido peso en la semana, son agrupados en grupos de 3 (tres),
preferentemente del mismo sexo, y acomodados en boxes apropiados, instalados en
un ambiente libre de perturbaciones de cualquier especie que puedan excitarlos
y a una temperatura que no presente diferencias de +/- 2 °C con relación a la temperatura del ambiente donde se encuentran alojados los animales (20 - 23 °C), suspendiéndose su alimentación 12 horas antes del inicio del ensayo, pudiendo mientras tanto
tener acceso al agua.
e) "Registro de la temperatura: Utilizar un
termómetro clínico o cualquier otro sensor de temperatura calibrado para
asegurar una precisión de +/- 0,1 °C y que haya sido probado para determinar
que la lectura máxima se alcanza en menos de 5 minutos. El sensor de
temperatura se deberá introducir en el recto de animal en ensayo hasta una
profundidad de no menos de 7,5 cm. Si el sensor debe permanecer en el recto
durante todo el período de registro, se debe inmovilizar al conejo mediante un
cepo holgado que le permita adoptar una postura natural. Cuando se emplea un
termómetro clínico, dejar transcurrir el tiempo necesario (previamente
determinado) para que alcance la temperatura máxima, antes de proceder a la
lectura.
f) Los animales seleccionados para el ensayo deben
presentar una temperatura corporal no superior a 39,8 °C, procediéndose a tomar 2 medidas de la temperatura, 30 y 60 minutos antes de la realización
del ensayo, para verificar la constancia de la temperatura.
g) Los animales de un mismo grupo de ensayo no deben
presentar una variación de temperatura de más de 1 °C de uno a otro.
h) Los animales que presenten variaciones de temperatura de +/- 0,2 °C, no deben emplearse en el ensayo de piretógenos.
i) La media de las temperaturas registradas se
considera la temperatura de control.
L-2.4. Muestreo y preparación de la muestra para el
ensayo.
Proceder
conforme a lo indicado en las monografías y en las normas específicas para
soluciones parenterales de gran volumen (SPGV).
L-2.5. Procedimiento para el ensayo
a) En condiciones asépticas, transferir la solución
en ensayo a jeringas y mantenerlas en estufa a 37 ºC hasta la hora de ensayo.
b) Para hacer resaltar mejor las venas auriculares y
al mismo tiempo promover su asepsia, frotarlas con un algodón embebido en
solución alcohólica.
c) Ajustar el volumen a ser inyectado de acuerdo con
el peso del animal (10 mL de solución en ensayo por kilogramo de peso corporal)
d) Inyectar la solución en ensayo por la vena
marginal de una de las orejas de los conejos, con un flujo de 3 mL por minuto.
e) Al finalizar la inyección, retirar la jeringa y
comprimir el lugar de la inyección con un algodón seco para obtener el sangrado
y evitar la formación de un hematoma.
f) Registrar la temperatura a 1 (una), 2 (dos) y 3
(tres) horas posteriores a la inyección. Un descenso de temperatura deberá ser
registrado como 0 (cero).
L-2.6. Interpretación del ensayo
a) La temperatura máxima registrada para cada conejo
es considerada como su respuesta. Cuando las temperaturas medidas luego de la
inyección fueran inferiores a la temperatura de control, la respuesta equivale a
una elevación de temperatura cero.
b) Si ninguno de los 3 (tres) conejos presentasen
elevaciones de temperatura de 0,5 ºC o más, sobre sus respectivas temperaturas
de control, el material en ensayo cumple los requisitos con relación a la
ausencia de piretógenos.
c) Si algún conejo presentara un aumento de
temperatura de 0,5 ºC o más, el ensayo deberá ser repetido usando 5 (cinco)
diferentes animales.
d) Si no más de 3 (tres) de los 8 (ocho) conejos
presentara elevaciones de temperatura de 0,5 ºC o más, y si la suma de las elevaciones de las temperaturas de los 8 (ocho) animales no excede los 3,3 ºC, el material en ensayo cumple los requisitos con relación a la ausencia de piretógenos.
L-3.
ENSAYO DE ENDOTOXINAS BACTERIANAS
L-3.1. Objetivo.
El siguiente
ensayo se establece para estimar la concentración de las endotoxinas
bacterianas que pueden estar presentes en la muestra del producto a analizar.
L-3.2. Generalidades.
Se utiliza para ello el reactivo
Lisado de Amebocitos de Limulus (LAL) que ha sido obtenido del lisado de
extractos acuosos de los amebocitos circulantes del cangrejo herradura,
"Limulus polyphemus", preparado y caracterizado para ser usado como
un reactivo de LAL, por formación de un gel.
La
determinación del punto final de la reacción se hace con diluciones a partir
del material en ensayo, en comparación directa con diluciones paralelas de una
endotoxina de referencia, y las cantidades de endotoxina se expresan en
Unidades de Endotoxina.
Alternativamente
podrán emplearse otros métodos. Los más conocidos son: turbidimétricos y
colorimétricos (incluyendo determinaciones cinéticas). Estos ensayos requieren
el establecimiento de una curva de regresión standard y el contenido de
endotoxina del material en ensayo, se determinan por interpolación en la misma.
Los
procedimientos incluyen la incubación durante un tiempo de reacción
preseleccionado de la endotoxina y las soluciones de control con reactivo LAL y
la lectura de la absorbancia espectrofotométrica de la luz a una longitud de
onda adecuada. En el caso del procedimiento turbidimétrico, la lectura se hace
inmediatamente al final del período de incubación o en la determinación
cinética (turbidimétrico y colorimétrico) la absorbancia se mide durante todo
el período de reacción y los valores de velocidad se determinan a partir de
estas lecturas.
En el
procedimiento colorimétrico de punto final, la reacción se detiene al final del
tiempo preseleccionado, por la adición, antes de la lectura, de una cantidad
apropiada de agente que detenga la reacción enzimática.
L-3.3. Estandares de referencia.
L-3.3.1. Endotoxina estandar de referencia (RSE).
La endotoxina standard de referencia
(RSE) es la endotoxina que tiene una potencia definida en Unidades de
Endotoxina (EU) por frasco, y que será reconstituida de acuerdo a las
instrucciones del rótulo. Se emplea este concentrado para efectuar las
diluciones seriadas adecuadas. Se conserva en heladera por no más de 14 días.
Se lleva a temperatura ambiente, si es necesario, y se agita en Vortex durante
no menos de 5 min. antes de usar. Se agita cada dilución en Vortex durante no
menos de 1 min. antes de efectuar la siguiente dilución. No se deben almacenar
las diluciones.
L-3.3.2. Endotoxina standard de control (CSE).
Una endotoxina standard de control
(CSE) es una preparación de endotoxina distinta a la RSE que ha sido estandarizada contra la RSE. Si una CSE es una preparación aún no
caracterizada adecuadamente, su evaluación debe incluir, parámetros que la
caractericen tanto por calidad de endotoxina y comportamiento (tal como
reacción en el conejo) cuanto por adecuabilidad del material para servir como
una referencia (tal como uniformidad y estabilidad).
Se deben incluir procedimientos
detallados para sus pesadas y/o reconstitución y uso para asegurar uniformidad
en el comportamiento.
La estandarización de una CSE contra la RSE usando un reactivo LAL y el procedimiento de coagulación en tubo se pueden efectuar
ensayando un mínimo de 1 frasco de la CSE y un frasco de la RSE como se señala en "Procedimiento para el Ensayo", pero usando los tubos
por cuadruplicados en cada nivel de la serie de dilución para la RSE y cuadruplicados similarmente para cada frasco.
El antilogaritmo de la diferencia
entre la media del log10 del punto final de la RSE y la media del log10 del punto final de la CSE es la potencia estandarizada de la CSE que entonces se debe convertir y expresar en Unidades/nanogramo, bajo
condiciones de secado establecidas para la CSE, o en unidades por frasco, lo que sea más apropiado. Se standariza cada lote nuevo de CSE antes de su uso en
el ensayo.
La calibración de una CSE en términos
de la RSE debe hacerse con el lote específico de Reactivo LAL y el
procedimiento de ensayo con el cual se va a usar.
Una CSE adecuada tiene una potencia de
no menos de 2 Unidades de Endotoxina por nanogramo y no más de 50 Unidades por
nanogramo cuando se encuentra como granel, bajo condiciones uniformes de secado
establecidas.
L-3.4. Ensayos preparatorios.
Confirmar la sensibilidad rotulada del reactivo LAL,
a utilizar.
Tratar los
recipientes y utensilios a emplear de modo de destruir endotoxinas extrañas
superficiales que puedan estar presentes, por ejemplo, calentándolos en una
estufa a 250 ºC durante 30 minutos o 180 ºC durante 3 horas.
La validez de
los resultados de los ensayos para endotoxinas bacterianas requiere una
demostración adecuada de que las muestras del producto o de soluciones, lavados
o extractos de estos a los cuales se va a aplicar el ensayo, no inhiban o
intensifiquen la reacción o interfieran de cualquier otra manera en el ensayo.
La validación se realiza por el ensayo de inhibición o intensificación,
incluyendo controles negativos apropiados. Se debe repetir la validación se
cambia la fuente del reactivo LAL o el método de fabricación o la formulación
del producto.
L-3.4.1. Ensayo de Sensibilidad.
Se debe confirmar la sensibilidad del
reactivo LAL usando 1 frasco por lote y 1 frasco de endotoxina. Preparar una
serie de diluciones de Endotoxina (RSE o CSE) con concentraciones de 0,25l
, 0,5l
, 1
l, 2l
, en cuadruplicado, donde l es la sensibilidad rotulada en el Reactivo LAL en
EU/ml. Incluir los controles negativos. La media geométrica de las
concentraciones en el punto final (ver "Cálculo e interpretación")
debe ser mayor o igual a 0,5
l y menor o igual a 2l
.
L-3.4.2. Ensayo de Inhibición o intensificación.
Llévese a cabo el ensayo sobre
alícuotas de la muestra o sobre una dilución que no exceda la máxima dilución
válida, en las cuales no exista endotoxina detectable. Llévese a cabo el
ensayo, sobre la muestra sin adición y con adición de endotoxina, para dar
contracciones finales de 2.0
l, 1.0l
, 0.5
l, 0,25l
, como se indica bajo Procedimiento
para el Ensayo, pero usando no menos de cuatro réplicas por muestra sin
adición y con adición de endotoxina.
En paralelo ensáyense por duplicado
las mismas concentraciones de endotoxina en agua y también controles negativos
no tratados. Calcúlese la media geométrica del punto final de la concentración
de endotoxina para la muestra como se describe en Cálculo e Interpretación.
El ensayo es válido para el producto si la media geométrica del punto final de
la concentración de la muestra es mayor o igual que 0.5
l y menor o igual que 2.0l
.
Si el resultado obtenido para las
muestras a las cuales se les ha agregado endotoxina está fuera del límite
especificado, el producto es inadecuado para el ensayo de endotoxinas
bacterianas. En el caso de inyecciones o soluciones para administración
parenteral, se pueden adecuar diluyéndolas apropiadamente.
Repítase el ensayo para inhibición o
intensificación usando las muestras diluidas por un factor que no exceda la Máxima dilución válida.
Para subsecuentes determinaciones de
endotoxina en las muestras de ensayo se emplea la dilución que no exceda la
máxima dilución válida y sea suficiente para superar la inhibición o
intensificación. Si bajo las condiciones del ensayo de inhibición o
intensificación son detectables endotoxinas endógenas en las muestras no
tratadas, las mismas pueden adecuarse removiendo la endotoxina presente por
ultrafiltración o por una dilución apropiada.
Dilúyase la muestra no tratada (como
se indica, cuando sea factible, para su administración o uso), a un nivel que
no exceda la máxima dilución válida a la cual ninguna endotoxina es detectable.
Se repite el ensayo de inhibición o
intensificación usando la muestra a éstas dilusiones.
L-3.4.3. Máxima Dilución Válida (MVD).
La máxima dilución válida es apropiada
a) para inyectables.
b) para soluciones para administración parenteral en
la forma reconstituida para administración.
c) cuando sea aplicable, a la cantidad de droga por
peso, si el volumen del producto pudiera variar en su dosificación.
Cuando se especifique la concentración
límite de endotoxina en la monografía individual en términos de volumen (en
EU/ml), se divide el límite por
l que es la sensibilidad indicada en la etiqueta (en
EU/ml) del listado empleado en el ensayo, para obtener el factor MVD.
Cuando la concentración límite de
endotoxina se especifica en términos de masa de las unidades de droga activa
(en EU/mg ó en EU/unidad), se multiplica el límite por la concentración (en
mg/ml ó u/ml) de la droga en la solución ensayada, o de la droga reconstituida
de acuerdo con las instrucciones que figuran en la etiqueta y se divide el
producto de la multiplicación por l
, para obtener el factor MVD. El
factor MVD así obtenido es el factor de dilución límite para la preparación
para que el ensayo sea válido.
L.3.5. Procedimiento para el ensayo.
Al preparar y
realizar el ensayo, se deben tomar precauciones al manipular las muestras para
evitar una contaminación bacteriana grosera.
Para validar
el ensayo para un producto, o determinaciones de límite de endotoxina, o para
propósitos especiales done se especifique, el ensayo de las muestras se realiza
cuantitativamente para determinar puntos finales de respuesta por lecturas de
gel coagulado. Se hacen diversas diluciones de la muestra y de la endotoxina
standard, se seleccionan las diluciones de modo que correspondan a una serie
geométrica en la cual cada una de ellas es más grande que la más próxima
inferior en una relación constante. A menos que se tengan datos que lo
permitan, no hay que almacenar endotoxinas diluidas porque pierden actividad
por absorción. En el ensayo deben incorporarse controles negativos y positivos.
Para cada nivel de la serie de diluciones de la muestra en ensayo debe operarse
por lo menos por duplicado. Sea que el ensayo se emplea como un ensayo límite o
como una determinación cuantitativa, se debe realizar en paralelo una serie
duplicada de tubos de reacción de dilución de endotoxina standard. Cada grupo
de tubos de reacción debe incluir una serie de dilución de la endotoxina
standard, la que deberá ser incubada simultáneamente en las mismas condiciones
ambientales.
L-3.5.1. Preparación.
Puesto que la forma y cantidad por
frasco de la endotoxina standard y el reactivo LAL puede variar, la
reconstitución y/o dilución del contenido deberá efectuarse como se indique en
el rótulo.
El pH de la mezcla de la muestra y del
reactivo LAL deberá estar entre 6,0 a 7,5 a menos que se indique otra cosa.
Se puede ajustar el pH por la adición
de hidróxido de sodio o ácido clorhídrico, o reguladores adecuados, libres de
endotoxina.
L-3.5.2. Procedimiento.
A tubos de ensayo de 10 mm x 75 mm se les agregan alícuotas del reactivo LAL reconstituido apropiadamente y los volúmenes
especificados de muestras, standard de endotoxina, controles negativos y un
control positivo del producto que consisten en el producto, o soluciones o
lavados o extractos de éstos a los cuales la RSE (o una CSE estandarizada) ha sido agregada a una concentración de endotoxina de 2
l
para aquel reactivo LAL. Ver ensayo de confirmación de la sensibilidad del
reactivo LAL.
Se agita cada uno suavemente para
mezclar y se coloca en un dispositivo para incubar, tal como un baño de agua o
un block calefactor, registrando exactamente la hora. Se incuba cada tubo, sin
agitarlo, por 60 min +/- 2 min a 37+ºC +/- 1ºC y se retira cuidadosamente para su observación.
La reacción positiva se caracteriza
por la formación de un gel firme que se mantiene cuando se invierte el tubo a
180º. Se registra tal resultado como positivo (+).
Un resultado negativo se caracteriza
por la ausencia de tal gel o por la formación de un gel viscoso que no mantiene
su integridad. Se registra tal resultado como negativo (-).
Se manipulan los tubos con cuidado y se evita
someterlos a indeseadas vibraciones, porque de lo contrario pueden resultar
falsos negativos.
El ensayo no es válido si el control
positivo del producto da negativo, o el standard de endotoxina no muestra que
la concentración del punto final está dentro de +/- una dilución al medio a
partir de la sensibilidad indicada en la etiqueta del reactivo LAL, o si algún
control negativo da positivo.
L-3.6. Cálculo e interpretación.
L-3.6.1. Cálculo de la media geométrica.
El punto final es la última dilución
positiva en una serie de concentraciones decrecientes de endotoxinas, muestra o
muestra que se ha adicionado con endotoxina. Registrar la concentración (E) en
cada punto final, para cada serie replicada de diluciones.
Determinar el logaritmo de cada
concentración punto final, (e) y calcular la media geométrica de las
concentraciones punto final usando la siguiente fórmula;
Media geométrica de las
concentraciones punto final = antilog
(åe/f)
Donde åe
es la suma de los logaritmos de la serie de diluciones de la muestra punto
final y f es el número de réplicas por cada una.
L-3.6.2. Cálculo del contenido de endotoxina
Calcular la concentración de
endotoxina (en unidades por ml o en unidades por g o por mg) en el producto que
se ensaya por la fórmula:
l
U
l = sensibilidad impresa en el rótulo del listado
utilizado en el ensayo expresada en EU/ml
U = antilog (åe/f)
e = es el log10 de los
factores de diluciones del punto final, expresados en fracción decimal.
f = es el número de réplicas de tubos
de reacción leídos en el punto final para la muestra ensayada.
L-3.6.3. Interpretación.
El producto
cumple los requerimientos del ensayo cuando la concentración de endotoxina es
no mayor que la especificada en la monografía individual.
L-4.
ENSAYO ESTERILIDAD
L-4.1. Condiciones generales.
Los siguientes procedimientos son
aplicables para determinar si una Solución Parenteral de Gran Volumen
estéril cumple con los requerimientos establecidos en la monografía individual
con respecto al ensayo de Esterilidad.
Para el uso de los procedimientos del ensayo de
esterilidad como parte del control de calidad en el proceso de fabricación, ver
E-3.1.9. Esterilización y Seguridad de Esterilidad.
Considerando la posibilidad de que los
resultados positivos puedan deberse a técnicas asépticas defectuosas o a
contaminación ambiental en el ensayo, se incluyen algunas medidas en Interpretación
de los resultados de los Ensayos de Esterilidad para un ensayo de dos
etapas.
Pueden emplearse procedimientos
alternativos para demostrar que una Solución Parenteral de Gran Volumen
es estéril, asegurando que los resultados obtenidos son al menos de
confiabilidad equivalente.
En el caso de
una controversia, cuando se obtenga evidencia de contaminación microbiana por
el procedimiento dado en esta Norma, el resultado obtenido es concluyente de
que el producto no cumple con los requerimientos del ensayo.
En forma
similar, en caso de no evidenciar contaminación microbiana por el procedimiento
dado en esta Norma, el producto cumple con los requerimientos del ensayo.
Para información adicional, ver E-3.1.9.
Esterilización y Seguridad de Esterilidad
L-4.2. Medios de cultivo.
Los medios
para los ensayos pueden prepararse como se describe más adelante, o utilizar
mezclas deshidratadas que dan formulaciones similares que pueden usarse
considerando que, cuando se reconstituyen como lo indica el fabricante o
distribuidor, tienen propiedades de promoción del crecimiento iguales o
superiores a las obtenidas de las fórmulas citadas en esta Norma.
I. Medio Fluido de Tioglicolato
|
L -
Cistina.............................................................................................0.5
g
|
|
Cloruro de
Sodio..................................................................................2.5
g
|
|
Dextrosa
Monohidratada.....................................................................
5.5 g
|
|
Agar,
granulado (el contenido de humedad)
no excede
el 15%.................................................................................0.75
g
|
|
Extracto de
Levadura (soluble en agua) ..............................................5.0
g
|
|
Digesto
Pancreático de
Caseína..........................................................15.0 g
|
|
Tioglicolato
de Sodio...........................................................................
0.5 g
|
|
o Acido
Tioglicólico...............................................................................0.3
ml
|
|
Solución de
Resazurina Sódica (1 en 1000)
recientemente
reparada.......................................................................1.0
ml
|
|
Agua..................................................................................................
1000 ml
|
pH después de la esterilización: 7. 3
+/- 0.2
Mezclar y calentar hasta disolución.
Ajustar la solución con hidróxido de sodio 1 N de forma que, después de la
esterilización, tenga un pH de 7.1 +/-0.2. Filtrar mientas esté caliente por un
papel de filtro, si es necesario.
Colocar el medio en recipientes
adecuados, que ofrezcan una relación superficie – profundidad en el medio de
forma que no más que la mitad superior del medio sufra un cambio de color
indicativo de la captación de oxígeno al final del periodo de incubación.
Esterilización en autoclave. Si más
del tercio superior adquiere un color rosa, el medio se puede recuperar una vez
por calentamiento en un baño de vapor o con vapor fluente hasta que desaparezca
el color rosa. Cuando esté listo para su uos, no más del décimo superior del
medio debe tener un color rosa.
Usar Medio Fluido de Tioglicolato incubándolo
en condiciones aeróbicas.
II. Medio de Digesto de Semilla de
Soja - Caseína
|
Digesto pancreático de
Caseína.........................................
|
17.0 g
|
|
Digesto papaico de Harina de Semilla de Soja
..................
|
3.0 g
|
|
Cloruro de
Sodio..................................................................
|
5.0 g
|
|
Fostato de potasio
dibásico.................................................
|
2.5 g
|
|
Dextrosa Monohidratada
.....................................................
|
2.5 g
|
|
Agua
....................................................................................
|
1000 ml
|
PH después de la esterilización: 7.3
+/- 0.2
Disolver los sólidos en agua, calentar
ligeramente hasta disolución. Enfriar la solución a temperatura ambiente, y
ajustar con hidróxido de sodio 1 N, si es necesario, para obtener un pH de 7.3
+/-02 después de la esterilización. Filtrar, si es necesario, y colocarlo en
recipientes decuados. Esterilizar por vapor.
Usar Medio de Digesto de Semilla de
Soja – Caseína incubándolo bajo condiciones aeróbicas.
L-4.3. Ensayo de promoción de crecimiento.
Confirmar la
esterilidad de cada lote de medio por incubación de recipientes
representativos, a la temperatura y tiempo especificados en el ensayo.
Analizar cada
carga autoclavada de cada lote de medio sobre sus propiedades de promoción del
crecimiento inoculando separadamente recipientes de ensayo de cada medio por
duplicado con 10 a 100 microorganismos viables de cada una de las cepas que
figuran en la tabla adjunta, e incubando de acuerdo a las condiciones
especificadas.
Los medios de
ensayo son satisfactorios si aparece una clara evidencia de crecimiento en
todos los recipientes con medios inoculados dentro de los 7 días. Los ensayos
pueden realizarse simultáneamente con el uso de los medios de ensayo para
análisis de esterilidad. La prueba de esterilidad se considera no válida si el
medio de ensayo muestra respuesta de crecimiento inadecuada.
Si un medio
recientemente preparado no se usa dentro de los 2 días, conservarlo en la
oscuridad, preferiblemente entre 2º y 25º.
Los medios
terminados, si se conservan en recipientes no herméticos, pueden usarse no más
de un mes, asegurando que son analizados dentro de la semana de uso y que
cumplen los requerimientos de color del indicador. Se conserva en recipientes
cerrados adecuados, los medios pueden usarse durante no más de un año,
asegurando que se realiza un análisis de promoción de crecimiento cada tres
meses y si cumplen con los requerimientos de color del indicador.
|
Medio
|
Microorganismos de ensayo*
|
Incubación T (ºC)
|
|
Tioglicolato
fluido
|
(1) Bacillus
subtilis (ATCC Nº 6633)+
|
30 a 35
|
|
(2) Candida
albicans (ATCC Nº 10231)
|
30 a 35 A+
|
|
(3) Bacteroides
vulgatus (ATCC Nº 8482)++
|
30 a 35
|
|
Digesto Semilla de
soja-caseína
|
(1) Bacillus
subtilis (ATCC Nº 6633)+
|
20 a 25
|
|
(2) Candida
albicans (ATCC Nº 10231)
|
20 a 25 A+
|
T (ºC): temperatura (ºC)
A+: aeróbicas
*Del American Type Culture Collection, 12301
Parklawn Drive, Rockville, MD 20852.
Nota- Deben emplearse las técnicas de mantenimiento de
los lotes de cultivo para siembra de forma que los microorganismos viables
usados para la inoculación no tengan más de cinco repiques de los cultivos
ATCC.
+Si no se desea un organismo formador
de esporas, usar Micrococcus luteus (ATCC Nº. 9341) a las temperaturas
de incubación indicadas en la Tabla.
++ Si se desea un organismo formador
de esporas, usar Clostridium sporogenes (ATCC Nº 11437) a la temperatura
de incubación indicada en la Tabla.
L-4.4. Procedimiento para el ensayo.
Se utiliza la técnica de filtración
por membrana, empleando la cantidad de unidades, volumen de solución y medio de
cultivo, indicados en la tabla.
|
Tabla de Cantidades
|
|
Contenido (mL)
|
A
|
V M
|
Nº de envases/medio
|
|
100 a 500
|
contenido total
|
100
|
10
|
|
Más de 500
|
500 mL
|
100
|
10
|
V M: Volumen mínimo de cada medio.
A: Volumen mínimo tomado de cada envase por cada
medio.
L-4.4.1. Equipamiento.
Una unidad de filtración por membrana
adecuada consiste en un equipo que facilita el manipuleo aséptico de los
productos a analizar y que permite retirar la membrana procesada asépticamente
para la inoculación en el medio adecuado o un equipo donde puedan agregarse
medios estériles al filtro sellado e incubar la membrana in situ.
Una membrana generalmente aceptada
para la prueba de esterilidad tiene una porosidad nominal de 0.45+/-0,02 um, un
diámetro de aproximadamente 47 mm y una velocidad de flujo de 55 a 75 ml de agua por minuto a una presión de 70 cm de mercurio.
La unidad completa puede armarse y
esterilizarse con la(s) membrana(s) en su lugar antes de usarse en el ensayo, o
las membranas pueden esterilizarse separadamente por cualquier modo que
mantenga las características de comportamiento del filtro y asegure la
esterilidad del filtro y el equipo.
L-4.4.2. Procedimiento.
Transferir asépticamente los volúmenes
requeridos para ambos medios, como se indica en la tabla de cantidades, ya sea
directamente en uno o dos equipos de filtración por membrana separados, o en un
recipiente estéril para hacer un pool antes de transferirlo.
Si el volumen de líquido es de 100 ml
hasta 500 ml, transferir asépticamente el contenido completo de no menos de 10
envases a través de cada uno de los equipos de filtros, o no menos de 20
envases si se usa sólo un equipo de filtro. Si el volumen del líquido en el
producto es más de 500 ml, transferir asépticamente no menos de 500 ml, de cada
uno de no menos de 10 envases, a través de cada uno de los equipos, o no menos
de 20 envases si se usa sólo un equipo de filtro. Inmediatamente pasar cada
muestra a través del filtro con la ayuda de vacío o presión.
En algunos casos, cuando el líquido es
altamente viscoso y no rápidamente filtrable a través de una o dos membranas,
pueden necesitarse más de dos equipos de filtros, en esos casos, la mitad del
número de membranas usadas se incuban en cada medio, considerando que se cumpla
con los volúmenes y requerimientos para el número de envases por medio.
Si el producto es bacteriostático o
fungistático, enjuagar la(s) membrana(s) con tres porciones de 100 ml de Líquido
de lavado A.
Nota:
Líquido de lavado A
Disolver 1 g de Digesto Péptico de Tejido Animal (Peptona Bacteriológica), en cantidad suficiente de agua para
llevar a 1 litro, clarificar por filtración o centrifugado y ajustar el pH a
7,1 +/- 0,2. Fraccionar en cantidades de 100 ml y esterilizar por vapor.
Retirar asépticamente la(s)
membrana(s) del soporte(s), cortar la membrana por la mitad (si solo se usa
una), sumergir la membrana, o su mitad en 100 ml de Medio de Digesto de Semilla
de Soja-Caseína, e incubar entre 20º y 25º durante no menos de 7 días. En forma
similar, sumergir la otra membrana, o la otra mitad, en 100 ml de Medio Fluido
de Tioglicolato, e incubar entre 30º y 35º durante no menos de 7 días.
L-4.5. Interpretación de los resultados
L-4.5.1 Primer Paso.
En los intervalos indicados durante y
en la conclusión del período de incubación, examinar el contenido de todos los
recipientes para observar crecimiento microbiano, así como el desarrollo de
turbidez y/o crecimiento de superficie. Si no se observa crecimiento, el
producto analizado cumple con los requerimientos del ensayo de esterilidad.
Si se encuentra crecimiento, pero el
revisado del lugar donde se realizó el ensayo de esterilidad, de los materiales
usados, de los procedimientos de ensayo y de los controles negativos, indica
que pudo haber una técnica inadecuada o un mal manejo aséptico en el ensayo en
sí mismo, el Primer Paso es declarado no válido y debe repetirse.
Si se observa crecimiento microbiano pero no hay
evidencia que invalide el Primer Paso del ensayo, proceder con el Segundo Paso.
L-4.5.2. Segundo Paso.
El número mínimo de muestras elegidas
es el doble del número analizado en el Primer Paso. Los volúmenes mínimos
analizados de cada muestra y los medios y períodos de incubación son los mismos
que los indicados para el Primer Paso. Si no se encuentra crecimiento
microbiano, el producto analizado cumple los requerimientos del ensayo de
esterilidad. Si se encuentra crecimiento microbiano, el resultado así obtenido
es concluyente de que el producto analizado no cumple con los requerimientos
del ensayo de esterilida
Sin embargo, si puede demostrarse que el Segundo
Paso fue invalidado debido a una técnica aséptica defectuosa o inadecuada en la
realización del ensayo, el Segundo Paso puede repetirs
Nota: Toda vez que se encuentre crecimiento
microbiano, se procederá a su aislamiento e identificación. En caso de
encontrarse el mismo microorganismo en más de un ensayo, sin importar el paso
del análisis, se considerará que el producto analizado no cumple con los
requerimientos del Ensayo de Esterilida
L-5.
ENSAYOS DE REACTIVIDAD BIOLOGI
Se establecen
a continuación dos tipos de ensayos de reactividad biológica: "in
vitro" e "in vivo
El cumplimiento del ensayo "in vitro" exime de
la realización del ensayo "in vivo". Se podrá obvr el primero y
efectuar solamente el ensayo "in vivo".
L-5.1. Ensayo de reactividad biológica "in
vivo"
L-5.1.1. Condiciones generales.
El siguiente ensayo se emplea para
determinar la reactividad biológica de cultivos de células de mamíferos después
del contacto con plásticos elastoméricos y otros materiales poliméricos.
Es esencial disponer de la superficie específica
para extracción.
Cuando la superficie de la muestra no
pueda ser determinada, utilizar 0,1g de elastómero o 0,2g de material plástico
u otro material, por cada mL de líquido de extracción.
También es esencial tener cuidado en
la preparación de las muestras, para evitar contaminación con microorganismos u
otros materiales extraños.
L-5.1.2. Standardes de referencia.
Standard de referencia para controles negativos de
plásticos de USP.
Standard de referencia de sólidos de biorreacción
positiva USP.
Standard de referencia de extractos de biorreacción
positiva USP.
L-5.1.3. Preparación del cultivo celular.
Preparar múltiples cultivos de
fibroblastos de mamífero L-929 (línea celular ATCC CCL 1, Clon NCTC 929) en
medio esencial mínimo suplementado con suero, con una densidad de sembrado de,
aproximadamente, 10 células por mL. Incubar los cultivos a 37ºC +/- 1ºC durante no menos de 24 horas en una atmósfera de 5+/- 1% de dióxido de carbono en aire,
hasta obtener una monocapa, con una confluencia mayor del 80%.
Examinar los cultivos preparados bajo
un microscopio para asegurar monocapas uniformes, casi confluyentes.
(NOTA: la reproductibilidad de
los ensayos de reactividada biológica "in vitro" depende de la
obtención de una densidad de cultivo uniforme).
L-5.1.4. Solventes de extracción.
Usar solución inyectable de cloruro de
sodio al 0,9%. También pueden utilizarse medios de cultivo de células de
mamífero sin suero, o medios de cultivo de células de mamífero suplementados
con suero. Se usa medio suplementado con suero cuando la extracción se realiza
a 37ºC durante 24 horas.
L-5.1.5. Aparatos.
Autoclave. Emplear un autoclave capaz de mantener
una temperatura de 121ºC +/-2ºC, equipado con un termómetro, un manómetro, una
purga, una bandeja adecuada para acomodar los recipientes de prueba por encima
del nivel del agua, y un sistema de enfriamiento de agua que permitirá el
enfriamiento de los recipientes de ensayo hasta aproximadamente 20ºC, pero no por debajo de los 20ºC, inmediatamente después del ciclo de calentamiento.
Estufa. Utilizar una estufa, (preferiblemente un modelo de
convección mecánica), que mantendrá las temperaturas de operación en el rango
de los 50ºC a los 70ºC, dentro de los +/-2ºC.
Estufa de cultivo. Utilizar una estufa de cultivo capaz
de mantener una temperatura de 37ºC+/-1ºC y una atmósfera de 5+/-1% de dióxido de carbono en aire.
Nota: si se utilizan tubos con tapa, no es necesario
mantener una atmósfera de dióxido de carbono en la estufa de cultivo.
Envases de extracción.
Utilizar solamente envases, como ampollas
o tubos de cultivo con tapa a rosca o sus equivalentes, de vidrio Tipo I. Si se
usan tubos de cultivo o su equivalente, se cierran con la tapa a rosca con una
cubierta elastomérica adecuada. La superficie expuesta de la cubierta
elastomérica debe estar totalmente protegida con un disco sólido inerte de 50 a 75 um de espesor. Puede fabricarse un disco adecuado a partir de politetrafluroetileno.
Preparación del aparato.
Limpiar cuidadosamente todo el
material de vidrio con mezcla sulfocrómica y, si es necesario, con ácido
nítrico caliente, seguido de enjuague prolongado con agua estéril para
inyectables. Esterilizar los envases y equipos utilizados para la extracción,
transferencia o administración del material de prueba y secarlos por un proceso
adecuado. Si se usa óxido de etileno como agente esterilizante, dejarlos no
menos de 48 horas para una degasificación completa.
L-5.1.6. Procedimiento.
Preparación de la muestra para los extractos:
Seguir el procedimiento indicado en el
punto L-5. 2. 2 Preparación de la muestra.
Preparación de los extractos:
Seguir el procedimiento indicado en el
punto L-5. 2. 2. 3 Preparación del extracto, usando solución inyectable de
cloruro de sodio al 0,9% o medio de cultivo de células de mamífero sin suero
como solventes de extracción.
Nota: si la extracción se realiza a 37ºC durante 24 horas, en una estufa de cultivo, usar el medio de cultivo celular suplementado con
suero.
L-5.1.7. Ensayo de difusión en agar.
En este ensayo la capa de agar actúa
como un apoyo para proteger las células del daño mecánico, mientras que permite
la difusión de productos químicos extraíbles de las muestras poliméricas. Los
extractos de materiales que deben analizarse se aplican a un trozo de papel de
filtro.
L-5.1.7.1. Preparación de la muestra:
Usar extractos preparados en forma
directa o usar porciones de las muestras de ensayo que tengan superficies
chatas de no menos de 100 mm2 de superficie.
L-5.1.7.2. Procedimiento.
Preparar las monocapas en placas de 60 mm de diámetro usando 7 mL de Preparación de cultivo celular. Aspirar el medio de cultivo
de las monocapas y reemplazar con medio de cultivo suplementado con suero que
contenga no más del 2 % de agar.
Colocar las superficies chatas de la Preparación de la muestra, Control de plásticos negativo USP (para tener un
control negativo), y un Extracto de Biorreacción Positiva USP o un Sólido
Biorreacción Positiva USP (para tener un Control Positivo) en cultivos por
duplicado en contacto con la superficie de agar solidificada. Incubar todos los
cultivos durante no menos de 24 horas a 37ºC +/- 1ºC, preferiblemente en una estufa de cultivo humidificada conteniendo 5+/- 1% de dióxido de carbono.
Examinar en cada cultivo la reactividad alrededor de cada Muestra, Control
Negativo y Control Positivo, bajo microscopio, usando colorantes citoquímicos
si se desea.
L-5.1.8. Interpretación de los
resultados.
La reactividad biológica (degeneración
y malformación celular) se describe y califica en una escala de 0 a 4 (ver Tabla 1). Medir las respuestas obtenidas con el Control Negativo y el Control Positivo: El
ensayo es válido si la respuesta observada en los standardes de referencia
corresponde al grado de reactividad biológica señalado en cada uno de ellos.
Medir la respuesta obtenida de la Preparación de la Muestra.
La Muestra cumple con los requerimientos del ensayo si en
ninguno de los cultivos de células expuestos a la Muestra se observa más que una reactividad leve (Grado 2).
Repetir el ensayo si no se confirma su validez.
TABLA 1
GRADOS DE REACTIVIDAD PARA EL ENSAYO
DE DIFUSION EN AGAR
Grado
|
Reactividad
|
Descripción
de la zona de Reactividad
|
|
0
|
Ninguna
|
No hay zona
detectable alrededor de o debajo de la muestra.
|
|
1
|
Débil
|
Zona limitada al área debajo de la
muestra.
|
|
2
|
Leve
|
Zona que se
extiende menos de 0,5 cm más allá de la muestra.
|
|
3
|
Moderada
|
Zona que se
extiende de 0,5 a 1,0 cm más allá de la muestra.
|
|
4
|
Severa
|
Zona que se
extiende más de 1,0 cm desde la muestra, pero que no incluye al disco
completo.
|
L-5.2. Ensayo de reactividad "in vivo"
L-5.2.1. Condiciones generales.
Los siguientes ensayos se emplean para
determinar la respuesta biológica de animales frente a materiales plásticos y
otros materiales poliméricos utilizados en los envases de las SPGV a través de
la inyección de extractos preparados a partir del material en ensayo.
L-5.2.2. Preparación de la muestra
L-5.2.2.1. Cantidad de muestra
|
Espesor del material plástico (mm)
|
Cantidad de muestra para cada 20 mL de solución
extractiva
|
Subdivisión (mm)
|
|
<0,5
|
120 cm2
de superficie total (combinadas ambas caras)
|
50 x 3
|
|
0,5 a 1
|
60 cm2
de superficie total (combinadas ambas caras)
|
50 x 3
|
L-5.2.2.2. Soluciones extractivas.
Solución fisiológica para inyección
Solución: 1:20 de alcohol en solución fisiológica para inyección.
L-5.2.2.3. Preparación del extracto.
Colocar la muestra subdividida en un
frasco de vidrio limpio y estéril de 100 mL de capacidad. Lavar dos veces con
aproximadamente 70 mL de agua para inyectables, agitando por 30 segundos en
cada lavado y eliminando completamente el agua de lavado. Agregar al frasco de
vidrio conteniendo la muestra, 20 mL de solución extractiva. En paralelo,
preparar un control negativo del mismo modo, sin la muestra en ensayo. Para
cada solución extractiva requerida en el ensayo, preparar un extracto con la
muestra en ensayo y un extracto control negativo. Realizar la extracción por
calentamiento en autoclave a 121 ºC durante 60 minutos o en estufa a 70 ºC durante 24 horas, o a 50 ºC durante 72 horas. Las condiciones de extracción no deben causar
alteraciones físicas como fusión o aglutinamiento de los trozos material
plástico, pues esto provocaría una reducción del área superficial. Una leve
adherencia puede tolerarse. Enfriar a temperatura ambiente no inferior a 22 ºC. Agitar vigorosamente por algunos minutos y transferir, inmediatamente, cada extracto a un
recipiente seco y estéril en forma aséptica. Almacenar los extractos a una
temperatura entre 22 y 30 ºC, y utilizarlos dentro de las 24 horas como máximo.
L-5.2.3. Ensayo de inyección sistémica intravenosa
L-5.2.3.1. Procedimiento.
Utilizar ratones albinos sanos, no
utilizados anteriormente en ensayos, y con peso entre 17 y 23 gramos. Para cada grupo de ensayos utilizar ratones de un mismo origen. Proveer agua y alimento
apropiados para animales de laboratorio.
Inocular cada extracto y su
correspondiente control negativo en cada grupo de cinco ratones, en las
cantidades y vías de administración que se detallan a continuación:
|
Solución extractiva
|
Dosis/kilo de peso corporal
|
vía
|
Velocidad de inoculación (mL/seg)
|
|
Solución
fisiológica
|
50 mL
|
EV
|
0,1
|
|
Solución
1:20 de alcohol en solución fisiológica
|
50 mL
|
EV
|
0,1
|
Nota: EV = Endovenosa
L-5.2.3.2. Interpretación.
Después de la inoculación, observar a
los animales a las 4, 24, 48 y 72 horas. Si durante el período de observación,
ninguno de los animales inoculados con el extracto de la muestra presenta un
grado de reacción mayor que los animales inoculados con el extracto control, la
muestra se considerada satisfactoria en lo que respecta a este ensayo.
Si algún animal inyectado con el
extracto de la muestra presenta una leve señal de toxicidad, y no más de un
animal presenta graves síntomas de toxicidad o muerte, repetir el ensayo
utilizando grupos de 10 ratones cada uno.
Después de repetir el ensayo la
muestra se considera satisfactoria si ninguno de los animales inyectados con el
extracto de la muestra presenta un grado de reacción mayor que el observado en
los animales inyectados con el extracto control.
L-5.2.4. Ensayo intracutáneo.
L-5.2.4.1. Procedimiento.
Utilizar consejos albinos sanos, de
piel sensible y no utilizados anteriormente en ensayos. Los conejos deben ser
cuidadosamente rasurados el día del ensayo, a ambos lados de la columna
vertebral, y su piel debe estar libre de irritaciones o traumas.
Remover los pelos sueltos por medio de
aspiración y si es necesario, limpiar la piel con un hisopo de algodón embebido
con alcohol diluido al 70%, y dejar secar antes de iniciar la inoculación.
Agitar vigorosamente cada extracto
almacenado según lo descripto en la sección L.5 5.1, antes de preparar la dosis
a ser inoculada.
Inocular por vía intradérmica, 0,2 mL
de extracto de la muestra en cada uno de los 10 puntos de inoculación de un
lado de la columna vertebral, en dos conejos. Del mismo modo, inyectar 0,2 mL
del extracto control correspondiente en 5 puntos del otro lado de la columna
vertebral de esos mismos dos conejos.
L-5.2.4.2. Interpretación.
Examinar los lugares inoculados 24, 48
y 72 horas después de la inoculación para comprobar la presencia de una
reacción del tejido tal como eritema, edema o necrosis. Evitar tocar los puntos
de inoculación durante la manipulación u observación del animal.
Para facilitar la observación, pasar
solamente un algodón embebido con alcohol diluido al 70% sobre la piel del
animal.
Se clasifican las reacciones del
extracto de la muestra en relación con el extracto control de acuerdo con la
siguiente escala numérica:
Evaluaciones de las reacciones de la piel
|
Formación
de eritema o costra
|
Valor
|
|
ausencia de eritema
|
0
|
|
ligero
eritema casi imperceptible
|
1
|
|
eritema
bien definido
|
2
|
|
eritema moderado
o severo
|
3
|
|
eritema
severo (con ligera costra)
|
4
|
|
Formación
de edema
|
Valor
|
|
ausencia de
edema
|
0
|
|
edema casi imperceptible
|
1
|
|
edema
ligero con bordes bien definidos
|
2
|
|
edema moderado
(elevado aproximadamente 1 mm)
|
3
|
|
edema
severo (elevado más de 1 mm y extendido más allá del área de inoculación)
|
4
|
|
|
|
El resultado promedio del extracto de
la muestra no debe ser mayor que el resultado promedio del extracto control. En
caso que el promedio sea superior o igual, repetir el ensayo. Usar extracto
reciente en otros tres animales.
El criterio de aceptación es el mismo.
L-6. ENSAYO DE CONTAMINACION MICROBIANA
L-6.1. Consideraciones generales.
Este ensayo
se utiliza para determinar el número total de microorganismos y/o la presencia
de microorganismos indeseables en productos no estériles.
L-6.2. Recuento total de microorganismos aeróbicos
viables.
Determinar el
recuento total de aeróbicos viable por el método de filtración por membrana, el
método de recuento en placa o el método de dilución seriada según se determine.
Realizar la
determinación bajo condiciones diseñadas como para evitar la contaminación
accidental del producto a examinar. Las precauciones tomadas para evitar la
contaminación deben ser tales que no afecten ningún microorganismo que deba
revelarse en el ensayo.
A menos que
se establezca de otra manera, usar 10 g ó 10 ml del producto a examinar, tomados
con las precauciones referidas anteriormente. Para obtener la cantidad
requerida mezclar varias porciones elegidas al azar a partir del material a
granel o del contenido de un número suficiente de envases. Dependiendo de la
naturaleza del producto a examinar, diluir, disolver, suspender o emulsionar
usando un liquido adecuado. Eliminar cualquier propiedad antimicrobiana del
producto a examinar por dilución, neutralización o filtración.
Deben
utilizarse grados adecuados de dilución de forma que el número de unidades
formadoras de colonias esté dentro de los límites sugeridos para el método a
usar.
L-6.2.1. Preparación del producto.
Productos solubles en agua
Disolver o diluir 10 g ó 10 ml del producto a examinar en solución buffer cloruro de sodio-peptona pH 7.0 (L.6. 4. 1) u
otro líquido adecuado que demostrará no poseer actividad antimicrobiana en las
condiciones del test, y ajustar el volumen a 100 ml(2) con el mismo
líquido. Si es necesario, ajustar a aproximadamente pH 7.
(2) Las caracteristicas de algunos
productos pueden necesitar el uso de mayores volúmenes.
Productos no grasos insolubles en agua.
Suspender 10 g ó 10 ml del producto a examinar en solución buffer cloruro de sodio-peptona pH 7.0 u otro
líquido adecuado que haya demostrado no tener ninguna actividad antimicrobiana
en las condiciones del ensayo y diluir a 100 ml(2) con el mismo
líquido; si es necesario, dividir el producto a examinar y homogeneizar la
suspensión mecánicamente. Puede agregarse un agente tensioactivo adecuado como
0.1 por ciento m/V de polisorbato 80 para ayudar a la suspensión de sustancias
pobremente humectables. Si es necesario, ajustar la suspensión a
aproximadamente pH 7.
Productos grasos.
Homogeneizar 10 g o 10 ml del producto a examinar con 5 g de polisorbato 20 o polisorbato 80, calentado a no más
de 40ºC(1) si es necesario. Mezclar cuidadosamente mientras se
mantiene la temperatura en un baño de agua o en un horno. Agregar 85 ml de
solución buffer cloruro de sodio-peptona pH 7.0, calentada a no más de 40 ºC si es necesario. Mantener esta temperatura por el tiempo más corto necesario para la formación
de una emulsión y en cualquier caso durante no más de 30 minutos. Si es
necesario, ajustar la emulsión a aproximadamente pH 7.
(1)
Para algunos
productos puede ser necesario calentar a no más de 45°C durante el menor tiempo posible.
L-6.2.2. Procedimiento para el ensayo
L-6.2.2.1. Filtración por membrana.
Usar membranas filtrantes que tengan
un tamaño de poro nominal no mayor de 0,45 um y cuya efectividad para retener
bacterias haya sido establecida. Los filtros de nitrato de celulosa, por
ejemplo, se usan para soluciones acuosas, aceitosas y débilmente alcohólicas, y
los filtros de acetato de celulosa, por ejemplo, para soluciones fuertemente
alcohólicas.
El método que se describe considera
que se usan membranas de aproximadamente 50 mm de diámetro. Si se usan filtros de un diámetro diferente, los volúmenes de las diluciones y de lavado deben
ajustarse según corresponda. El aparato de filtración y la membrana deben ser
esterilizados por medios apropiados. El aparato debe diseñarse de forma que la
solución a examinar pueda introducirse y filtrarse bajo condiciones asépticas y
debe permitir la remoción de la membrana para luego transferirla al medio de
cultivo. Transferir 10 ml, o la cantidad de cada dilución representando 1 g del producto a examinar a cada una de dos membranas filtrantes y filtrar inmediatametne. Si es
necesario, diluir el producto preparado de forma que pueda esperarse un
recuento de colonias entre 10 a 100. Lavar cada membrana pasando a través del
filtro al menos tres porciones, cada una de aproximadamente 100 ml., de un
líquido adecuado como solución buffer de cloruro de sodio-peptona pH 7.0. Para
sustancias grasas, este líquido puede contener un agente tensioactivo como
polisorbato 20 o polisorbato 80. Transferir una de las membranas filtrantes,
destinada principalmente para la enumeración de las bacterias, a la superficie
de una placa de agar B y la otra, destinada principalmente para la enumeración
de hongos, a la superficie de una placa de agua C. Incubar la placa de agar B a
30°C - 35°C durante 5 días y la placa de agar C a 20°C - 25°C durante 5 días, a menos que se obtenga un recuento más confiable en un tiempo más corto.
Contar el número de colonias que se desarrollan. Calcular el número de
microorganismos por grano o por mililitro del producto a examinar, si es
necesario contar las bacterias y hongos separadamente.
L-6.2.2.2. Recuento en placa
Para Bacterias.
Usando placas de Petri de 9 cm a 10 cm de diámetro, agregar a cada placa una mezcla de 1 ml del producto ya preparado para su
examen en a) y aproximadamente 15 ml de agar B fundido, a no más de 45ºC. Como opción alternativa, distribuir el producto preparado sobre a superficie del medio
solidificado en una placa de Petri del mismo diámetro. Si es necesario, diluir
el producto preparado como se describió antes de forma que pueda esperarse un
recuento de colonias de no más de 300. Preparar al menos dos de estas placas de
Petri usando la misma dilución e incubar a 30ºC - 35ºC durante 5 días, a menos que se obtenga un recuento más confiable en un tiempo más corto. Contar el número
de colonias que se desarrollan. Calcular los resultados usando placas con el
mayor número de colonias pero considerando 300 colonias por placa como el
máximo concordante con una buena evaluación.
Para Hongos.
Usando placas de Petri de 9 cm a 10 cm de diámetro, agregar a cada placa una mezcla de 1 ml del producto ya preparado para su
examen en a) y aproximadamente 15 ml de agar C fundido, a no más de 45ºC. Como opción alternativa, distribuir el producto preparado sobre la superficie del medio
solidificado en una placa de Petri del mismo diámetro. Si es necesario, diluir
el producto preparado como se describió antes de forma que pueda esperarse un
recuento de colonias de no más de 100. Preparar al menos dos de esas placas
usando la misma dilución e incubar a 20ºC - 25ºC durante 5 días, a menos que se obtenga un recuento más confiable en un tiempo más corto. Contar las
colonias que se desarrollan. Calcular los resultados usando placas con no más
de 100 colonias.
L-6.2.2.3. Dilación Seriada.
Preparar una serie de doce tubos cada
uno conteniendo entre 9 ml y 10 ml de Calcio A. A cada uno de los primeros tres
tubos agregar 1 ml del producto diluido, disuelto u homogeneizado en la
proporción 1 en 10, como se describió antes. A los siguientes tres tubos,
agregar 1 ml de una dilución 1 en 100 del producto y a los siguientes tres
tubos agregar 1 ml de una dilución 1 en 1000 del producto. A los últimos tres
tubos agregar 1 ml del diluyente. Incubar los tubos a 30 ºC - 35ºC al menos durante 5 días. Los últimos tres tubos no deben mostrar crecimiento microbiano.
Si la lectura de los resultados es difícil o incierta debido a la naturaleza
del producto a examinar, subcultivar en un medio líquido o sólido y leer los
resultados después de otro período de incubación. Determinar el número más
probable de microorganismos por gramo o por mililitro del producto a examinar a
partir de la Tabla I.
TABLA I
Número más probable de microorganismos
|
Número de tubos donde se ve crecimiento
microbiano para cada cantidad de productos a examinar
|
Número más probable de microorganismos por gramo
o por milímetro
|
|
100 mg ó 0.1 mL por tubo
|
10 mg ó 0.01 mL por tubo
|
1 mg ó 0.001 mL por tubo
|
|
|
3
|
3
|
3
|
mayor de 1100
|
|
3
|
3
|
2
|
1100
|
|
3
|
3
|
1
|
500
|
|
3
|
3
|
0
|
200
|
|
3
|
2
|
3
|
290
|
|
3
|
2
|
2
|
210
|
|
3
|
2
|
1
|
150
|
|
3
|
2
|
0
|
90
|
|
3
|
1
|
3
|
160
|
|
3
|
1
|
2
|
120
|
|
3
|
1
|
1
|
70
|
|
3
|
1
|
0
|
40
|
|
3
|
0
|
3
|
95
|
|
3
|
0
|
2
|
60
|
|
3
|
0
|
1
|
40
|
|
3
|
0
|
0
|
23
|
|
Si para la primera columna el número de tubos que
muestra crecimiento microbiano es de dos o menos, el número más probable de
microorganismos por gramo o por mililitro es probablemente menor de 100.
|
L-6.2.3. Efectividad de los medios de cultivo y validez
de los métodos de recuento.
Cuando sea necesario, operar de la
siguiente forma: hacer crecer las siguientes cepas de ensayo separadamente en
tubos conteniendo Caldo A a 30°C - 35°C durante 18 horas a 24 horas o, para Candida
albicans, a 20°C - 25°C durante 48 horas.
Staphylococus aureus como ATCC 6538 P (PNCIB 8625, CIP
53.156) ó ATCC 6538 (NCIB 9518, CIP 4.83)
Bacillus subtilis
como ATCC 6633 (NCIB 8054, CIP 52.62)
Escherichia coli como ATCC 8739 (NCIB 8545, CIP 53.126)
Candida albicans como ATCC 2091 (CIP 1180.79) ó ATCC 10231 (NCPF
3179, CIP 48.72).
Diluir porciones de cada uno de los
cultivos usando solución buffer de cloruro de sodio-peptona pH 7.0 para hacer
suspensiones de ensayo conteniendo alrededor de 100 microorganismos viables por
mililitro.
Usar la suspensión de cada uno de los
microorganismos separadamente como control de los métodos de recuento, en
presencia y ausencia del producto a examinar si es necesario.
Cuando se ensaya el método, no debe
obtenerse, para cualquiera de los organismos de ensayo, un recuento que difiera
en más de un factor de 10 del valor calculado para el inóculo. Para ensayar la
esterilidad del medio y del diluyente y de la condición aséptica del ensayo,
realizar el método de recuento total de aeróbicos viables usando solución
buffer de cloruro de sodio-peptona pH 7.0 como preparación de ensayo. No debe
haber crecimiento de microorganismos.
L-6.2.4. Interpretación de los resultados.
De establecerse un límite, el mismo
estará indicado en la monografía individual correspondiente. Cuando el límite
se indique en forma exponencial deberá interpretarse de la siguiente manera:
102 microorganismos - límite máximo de
aceptación: 5 x 102 ,
103 microorganismos - límite máximo de
aceptación: 5 x 103 , etc.
L-6.3. Ensayos para la búsqueda de microorganismos
específicos
L-6.3.1. Enterobacterias y otras bacterias. Gram negativas
L-6.3.1.1.
Detección de
bacteria.
Preparar el producto a examinar como
se describe en L. 6. 2. 1, pero usando caldo D en lugar de solución buffer de
cloruro de sodio-peptona pH 7.0, homogeneizar e incubar a 35°C - 37°C, durante un tiempo suficiente para revivir las bacterias pero no tanto como para permitir
la multiplicación de los organismos (generalmente 2 h, pero no más de 5 h).
Agitar el envase, transferir la cantidad de contenido (homogenato a)
correspondiente a 1 g o 1 ml del producto a 100 ml de Caldo de enriquecimiento
e incubar a 35°C - 37°C durante 18 horas a 48 horas. Subcultivar en placas de
agar F. Incubar a 35°C - 37°C durante 18 horas a 24 horas. El producto pasa el
ensayo si no hay crecimiento de colonias de bacterias gram negativas en ninguna
placa.
L-6.3.1.2. Evaluación cuantitativa.
Inocular cantidades adecuadas de Caldo
de enriquecimiento E con el homogenato (a) y/o diluciones de éste, conteniendo
respectivamente 1.0 g, 0.1 g y 0.01 g ó 1.0 ml, 0.1 ml y 0.01 ml del producto a
examinar. Incubar a 35ºC - 37ºC durante 24 h a 48 h. Subcultivar cada uno de
los cultivos en una placa de agar F para obtener un aislamiento selectivo.
Incubar a 35ºC - 37ºC durante 18 h a 24 h. El crecimiento de colonias bien
desarrolladas, generalmente rojas a rojizas, de bacterias Gram negativas,
constituye un resultado positivo. Registrar la cantidad más pequeña de producto
que da un resultado positivo y la cantidad más grande que da un resultado
negativo. Determinar a partir de la Tabla II el número probable de bacterias.
Tabla II
Resultados para cada cantidad de producto
|
Número
probable de bacterias por gramo de producto
|
|
1.0 g ó 1,0 ml
|
0,1 g ó 0.1 ml
|
0.01 g ó 0.01 ml
|
|
|
+
|
+
|
+
|
más de 102
|
|
+
|
+
|
-
|
menos de 102
y más de 10
|
|
+
|
-
|
-
|
menos de 10
y más de 1
|
|
-
|
-
|
-
|
menos de 1
|
L-6.3.2 Escherichia coli.
Transferir la cantidad del cultivo en
Caldo D, preparado e incubado para el ensayo para enterobacterias y otras
bacterias Gram negativas, que represente 1 g ó 1 ml del producto inicial, a 100 ml de Caldo G, e incubar a 43°C - 45°C durante 18 h a 24 h. Subcultivar en
agar H e incubar a 43°C - 45°C durante 18 h a 24 h. El crecimiento de colonias
rojas, generalmente no mucoides de bastoncitos Gram negativo, a veces rodeadas
por una zona de precipitación rojiza, indica la posible presencia de E. coli.
Esto puede confirmarse por la formación de indol a 44 ± 0.5°C y por otras reacciones bioquímicas. El producto pasa el ensayo si esas colonias no se ven o si
las reacciones bioquímicas confirmatorias son negativas.
L-6.3.3. Salmonella.
Incubar una cantidad que represente 10 g ó 10 ml del producto, en Caldo D a 35°C - 37°C durante 5 h a 24 h, según corresponda para el
enriquecimiento. Transferir 10 ml del cultivo de enriquecimiento a 100 ml de
Caldo I e incubar a 42ºC - 43ºC durante 18 a 24 h. Subcultivar en al menos dos diferentes medios de agar elegidos entre de agar J, agar K y agar L. Incubar a 35°C -37°C durante 24 h a 48 h. La probable presencia de salmonelas está indicada por el crecimiento
de cultivos que tienen el siguiente aspecto:
agar J:
colonias bien desarrolladas, incoloras,
agar K:
colonias bien desarrolladas, rojas, con o sin
centros negros,
agar L:
colonias pequeñas, transparentes,
incoloras o rosas a blanco opaco, generalmente rodeadas de una zona rosa o
roja.
Transferir separadamente algunas de
las colonias en sospecha a un agar M en tubos, usando inoculación de superficie
y profunda. La presencia de salmonelas es provisionalmente confirmada si en una
inoculación profunda pero no en el cultivo de superficie hay un cambio de color
desde el rojo al amarillo y generalmente formación de gas, con o sin producción
de sulfuro de hidrógeno en el agar. La confirmación precisa puede realizarse
por los ensayos bioquímicos y serológicos adecuados. El producto pasa el ensayo
si los cultivos del tipo descripto no aparecen o si los ensayos confirmatorios
bioquímicos y serológicos son negativos.
L-6.3.4. Pseudomona aeruginosa.
Inocular 100 ml de Caldo A con 10 ml
del producto preparado para su examen en L-6. 2. 1 (preparación del producto),
o la cantidad del mismo que represente 1 g ó 1 ml del producto inicial. Mezclar e incubar a 35 °C - 37°C durante 24 h a 48 h. Subcultivar en una placa de
agar N e incubar a 35 °C - 37°C durante 24 h a 48 h. Si no se detecta
crecimiento de microorganismos, el producto pasa el ensayo. Si ocurre
crecimiento de colonias de bastoncillos Gram negativos, generalmente con una
fluorescencia verdosa, realizar un ensayo de la oxidasa y analizar el
crecimiento en Caldo A a 42°C. El producto pasa el ensayo si los cultivos del
tipo descripto no aparecen o si el ensayo bioquímico confirmatorio es negativo.
L-6.3.5. Staphylococcus aureus.
Preparar un cultivo de enriquecimiento
como se describió para Ps. aureginosa. Subcultivar en un medio adecuado
como placas de agar O. Incubar a 35°C - 37°C durante 24 h a 48 h. Si no se detectó crecimiento de microorganismos, el producto pasa el test. Colonias negras
de cocos Gram positivos generalmente rodeadas de zonas claras pueden iniciar la
presencia de S. aureus. Para los cocos catalasa positivos, la
confirmación puede efectuarse, por ejemplo, por los ensayos de coagulasa y
desoxiribonucleasa. El producto pasa el ensayo si los cultivos del tipo
descripto no aparecen o si los ensayos bioquímicos confirmatorios son
negativos.
L-6.3.6 Propiedades nutritivas y selectivas de los
medios y validez del ensayo para microorganismos específicos.
Cuando sea necesario, operar de la
siguiente manera: hacer crecer las siguientes cepas de ensayo separadamente en
tubos que contienen los medios indicados a 30°C - 35°C durante 18 h a 24 h.
Staphylococcus aureus como ATCC 6538 P Caldo A (NCIB 8625,
CIP 53.156) o ATCC6538 (NCIB 9518 CIP 4.83)
Pseudomonas aeruginosa como ATCC 9027 Caldo A (NCIB 8626,
CIP 82.118)
Escherichia coli como ATCC 8739 Caldo D (NCIB 8545, CIP
53.126)
Salmonella typhimurium(1) Caldo D
(1) No se recomienda número de cepa. Puede usarse
también una salmonella no patógena para el hombre, como Salmonella Abony (NCTC
6017, CIT 80.39)
Diluir porciones de cada uno de los
cultivos usando solución buffer de cloruro de sodio-peptona pH 7.0 para hacer
suspensiones de ensayo que contengan aproximadamente 1000 microorganismos
viable por mililitro. Mezclar volúmenes iguales de cada suspensión y usar 0.4
ml (aproximadamente 100 microorganismos de cada cepa) como un inóculo en los
ensayos para E. coli, salmonellas, Ps. aeruginosa y S. aureus, en presencia y
ausencia del producto a examinar si es necesario. Cuando se ensaya el método,
debe obtenerse un resultado positivo para el respectivo microorganismo.
L-6.4. Solución y medios de cultivo recomendados.
La siguiente
solución y medios de cultivo se encontraron satisfactorios a los efectos para
los cuales se recomiendan en el ensayo para contaminación microbiana. Pueden
utilizarse otros medios si tienen similares propiedades nutritivas y selectivas
para los microorganismos a analizar.
L-6.4.1. Solución bufferada cloruro de sodio-peptona pH
7.0
|
Fosfato diácido
de potasio
|
3.56 g Equivalente
|
|
Fosfato disódico
dihidratado
|
7.23 g a 0.067 M
|
|
Cloruro de sodio
|
4.30 g
|
|
Peptona (cárnea
o caseína)
|
1.0 g
|
|
Agua purificada
|
1000 mL
|
|
Puede
agregarse 0.1 por ciento m/V a 1.0 por ciento m/V de polisorbato 20 u 80.
Esterilizar por calentamiento en un autoclave a 121ºC durante 15 minutos.
|
L-6.4.2. Caldo A (medio digesto de caseína semilla de
soja)
|
Digesto
pancreático de caseína
|
17.0 g
|
|
Digesto
papaico de semilla de soja
|
3.0 g
|
|
Cloruro de
sodio
|
5.0 g
|
|
Fosfato
dipotásico
|
2.5 g
|
|
Dextrosa
monohidratada
|
2.5 g
|
|
Agua
purificada
|
1000 mL
|
|
Ajustar el
pH de forma que después de la esterilización sea de 7.3 + 0.2.
Esterilizar por calor en un autoclave a 121 °C durante 15 minutos.
|
L-6.4.3. Agar B (agar digesto de caseína semilla de
soja)
|
Digesto
pancreático de caseína
|
15.0 g
|
|
Digesto
pancreático de semilla de soja
|
5.0 g
|
|
Cloruro de
Sodio
|
5.0 g
|
|
Agar
|
15.0 g
|
|
Agua
purificada
|
1000 mL
|
|
Ajustar el pH de forma que después de la
esterilización sea de 7.3 ± 0.2. Esterilizar por calor en un autoclave a 121ºC durante 15 minutos.
|
L-6.4.4. C (Agar Sabouraud-dextrosa con
antibióticos)
|
Peptonas
(cárneas y de caseína)
|
10.0 g
|
|
Dextrosa monohidrato
|
40.0 g
|
|
Agar
|
15.0 g
|
|
Agua
purificada
|
1000 mL
|
|
Ajustar el pH de forma que después de
la esterilización sea de 5.6 ± 0.2. Esterilizar por calor en un autoclave a 121°C durante 15 minutos. Inmediatamente antes de su uso agregar 0.10 g de benzilpenicilina sódica y 0.10 g de tetraciclina por litro de medio como soluciones
estériles i, alternativamente, agregar 50 mg de cloranfenicol por litro de
medio antes de la esterilización.
|