RE/17/2016/GMC
FARMACOPEA
MERCOSUR: MÉTODOS DE FARMACOGNOSIA
VISTO: El Tratado de
Asunción, el Protocolo de Ouro Preto y las Resoluciones N° 31/11 y 22/14 del
Grupo Mercado Común.
CONSIDERANDO:
Que la Farmacopea MERCOSUR
tiene como objetivo establecer los requisitos mínimos de calidad y seguridad de
los insumos para la salud, especialmente de los medicamentos, apoyando las
acciones de reglamentación sanitaria y promoviendo el desarrollo técnico, científico
y tecnológico regional.
Que las especificaciones
farmacopeicas establecen, por medio de monografías, requisitos mínimos para el
control de seguridad y calidad de los insumos, especialidades farmacéuticas,
plantas medicinales y derivados producidos o utilizados en los Estados Partes.
Que las especificaciones
farmacopeicas son utilizadas como parámetro para las acciones de vigilancia
sanitaria, incluyendo el registro de medicamentos, inspecciones y análisis de
laboratorio.
Que la Farmacopea MERCOSUR
y la producción de patrones propios de calidad favorecen al desarrollo
científico y tecnológico de los Estados Partes, contribuyendo a la disminución
de la dependencia de proveedores extranjeros y promoviendo a la industria
regional.
Que la Farmacopea MERCOSUR
debe ser primordialmente sanitaria, con énfasis en la salud pública, y
presentar una metodología analítica accesible a los Estados Partes, buscando su
reconocimiento y respetabilidad internacional.
Que el diálogo regulatorio
y la integración entre los Estados Partes promueven el acceso de la población a
medicamentos con mayor calidad y seguridad.
Que el Acuerdo Nº 08/11 de
la Reunión de Ministros de Salud del MERCOSUR constituye un marco de referencia
para la Farmacopea MERCOSUR.
EL GRUPO MERCADO COMÚN
RESUELVE:
Art. 1 - Aprobar, en el
marco de lo establecido en la Resolución GMC N° 22/14, el método general
“Farmacopea MERCOSUR: Métodos de Farmacognosia”, que consta como Anexo y forma
parte de la presente Resolución.
Art. 2 - Los Estados
Partes indicarán en el ámbito del SGT N° 11 los organismos nacionales
competentes para la implementación de la presente Resolución.
Art. 3 - Esta Resolución
deberá ser incorporada al ordenamiento jurídico de los Estados Partes antes del
15/XII/2016.
CII GMC – Montevideo,
15/VI/16.
ANEXO
MÉTODOS DE FARMACOGNOSIA
DEFINICIONES
Drogas de origen natural
Son plantas,
cianobacterias, algas, hongos, líquenes, animales, que contengan sustancias o
grupos de sustancias responsables de una acción terapéutica. La droga se define
por la parte usada y el nombre científico (especie, variedad cuando aplique y
sigla del/los autor/es).
Droga vegetal
Plantas enteras y/o sus
partes, generalmente secas, no procesadas, pudiendo estar fragmentadas. También
se incluyen exudados (gomas, resinas, mucílagos, látex y ceras) que no hayan
sido sometidos a un tratamiento específico.
Preparado de droga vegetal
Preparados obtenidos
sometiendo las drogas vegetales a tratamientos tales como molienda, extracción,
destilación, prensado, fraccionamiento, purificación, concentración o
fermentación (tinturas, extractos, aceites fijos o volátiles, jugos y exudados
procesados).
MÉTODOS DE FARMACOGNOSIA
MUESTREO
Debido a las
características de las drogas vegetales, en particular su falta de
homogeneidad, se requieren procedimientos especiales en relación a los ensayos
a realizar.
Los procedimientos de
muestreo tienen en consideración tres aspectos: (a) número de envases que
contienen la droga; (b) grado de división de la droga y (c) cantidad de droga
disponible.
NÚMERO DE ENVASES
Examinar la integridad de
los recipientes de envase y la naturaleza de la droga contenida en ellos. Si el
examen externo de los envases y rótulos indica que puede considerarse el lote
como homogéneo, tomar muestras individuales de un número de envases
seleccionados aleatoriamente conforme se especifica en la Tabla 1. Si el lote
no puede considerarse homogéneo, fraccionarlo en sublotes que sean lo más
homogéneos posible y realizar el muestreo con cada uno como un lote.
Tabla 1. Número de envases
a muestrear
Número de envases Número
de envases a ser muestreados
1 a 3 Todos
4 a 10 3
11 a 20 5
21 a 50 6
51 a 80 8
81 a 100 10
Mas de 100 10%
GRADO DE DIVISIÓN Y
CANTIDAD DE DROGA
Tomar las muestras de las
secciones superior, media e inferior de cada envase. Recoger las muestras de
arriba hacia abajo y de abajo hacia arriba (dirección vertical) y lateralmente
(dirección horizontal).
Fragmentos inferiores a 1
cm
En el caso de los polvos o
material compuesto por fragmentos de menos de 1 cm, retirar la muestra a través
de un aparato de muestreo (tubo provisto de un dispositivo de cierre en la
base). Tomar muestras de mínimo 250g para lotes de hasta 100 kg de droga. Para
lotes mayores a 100 kg, tomar muestras de 250 g por cada 100 kg de droga y
componer una muestra final por cuarteamiento de 250 g.
Fragmentos superiores a 1
cm
Para drogas con
dimensiones superiores a 1 cm, retirar las muestras manualmente. Mezclar las
muestras retiradas de cada envase abierto, tomando la precaución de no aumentar
el grado de fragmentación o modificar significativamente el contenido de
humedad durante la manipulación.
Para cantidades de drogas
hasta 100 Kg, la muestra debe estar constituida por no menos de 500 g. Cuando
haya más de 100 Kg de droga, proceder al muestreo seguido de una selección por
cuarteo, generando muestra de 500 g al final del proceso.
Observaciones
En ambos casos, para
drogas con dimensiones inferiores o superiores a 1 cm, es posible muestrear
cantidades inferiores a las especificadas anteriormente, siempre que la
cantidad de droga disponible sea inferior a 10 Kg. Sin embargo, la muestra
final no deberá ser inferior a 125 g.
En caso de bultos o
envases grandes, las muestras deben ser recogidas a más de 10 cm de los bordes,
debido a que el contenido de humedad superficial puede ser diferente en las
capas más internas.
Cuarteo
Combinar y mezclar las
muestras tomadas de cada envase abierto, evitando aumentar el grado de
fragmentación o modificar significativamente el contenido de humedad durante la
manipulación.
Distribuir homogéneamente
la muestra tomada en forma de cuadrado y fraccionarla
en cuatro partes iguales.
Juntar dos partes opuestas y mezclarlas cuidadosamente. Juntar las dos
porciones restantes y repetir el procedimiento, si fuera necesario, hasta
obtener la cantidad requerida. Si existe diferencia acentuada en las
dimensiones de los fragmentos, realizar la separación manual y tomar nota de
los porcentajes aproximados de componentes de diferentes grados de división
encontrados en la muestra.
EXAMEN SENSORIAL Y
MICROSCÓPICO DE DROGAS VEGETALES
La identidad, pureza y
calidad de un material vegetal deben ser establecidas mediante examen visual
detallado, macroscópico y microscópico. Siempre que sea posible, el material
vegetal debe ser comparado con materia prima de referencia, o derivada de
muestra perfectamente identificada por Farmacopeas. Una muestra que no es
semejante en color, consistencia y olor debe ser descartada por no presentar
los requisitos mínimos especificados en las monografías. La identificación
macroscópica de las drogas, cuando se encuentran enteras, se basa en la forma,
tamaño, color, superficie, textura, fractura y apariencia de la superficie de
fractura. En virtud de que estas observaciones son subjetivas y podrían existir
adulterantes muy parecidos, es necesario realizar al mismo tiempo, el análisis
microscópico y físico-químico de la muestra. La observación microscópica es
indispensable cuando el material se encuentre triturado o en polvo.
Tamaño
Las medidas de longitud,
ancho y grosor deben coincidir con aquellas especificadas en las monografías.
Frutos y semillas pequeños exigen una muestra de diez (10) unidades y realizar
luego cálculos de media y desvío estándar.
Color
Examinar la muestra antes
de cualquier tratamiento, a la luz del día o sobre una lámpara de longitud de
onda similar a la luz natural. El color de la muestra debe ser comparado con el
material de referencia.
Superficie, textura y
fractura
Examinar la muestra antes
de cualquier tratamiento. Cuando sea necesario, utilizar un lente de 5x a 10 x.
Cuando sea indicado en la monografía, humedecer con agua o con el reactivo
especificado, para observar las características de la superficie de fractura.
Tocar el material para verificar si es blando o duro, doblar y partir el
material para obtener información sobre la fragilidad y apariencia de la
fractura, si es fibrosa, lisa, rugosa, granulada, entre otras.
Olor
Antes de verificar el olor
del material, asegurarse de que no existe riesgo de salud. Colocar una pequeña
muestra en la palma de la mano o en un recipiente de vidrio e inhalar
lentamente repetidas veces. Si el olor fuera indistinto, presionar parte del
material entre los dedos e inhalar nuevamente. Cuando la monografía indique que
se trata de material tóxico, colocar un poco de material triturado en agua
caliente. En primer lugar, determinar la intensidad del olor: ninguno, débil,
distinto o fuerte y, a continuación, la sensación causada por el olor:
aromático, frutal, enmohecido o rancio. Cuando sea posible, es importante
comparar el olor con una sustancia definida, como por ejemplo, la menta debe
tener olor similar al mentol y el clavo de olor, similar al eugenol.
Sabor
Testear el sabor solo
cuando sea exigido en la monografía.
Preparación del material
para el análisis microscópico
Hidratación o
ablandamiento del material - Los órganos vegetales o sus partes normalmente se
presentan secos, y para realizar los cortes y observaciones con un microscopio
óptico, es conveniente primero ablandarlos mediante tratamiento con agua
caliente o solución de hidratación. El tiempo necesario para el ablandamiento
de cada órgano vegetal o sus partes varía de acuerdo a su textura. Cuando se
trate de órganos frescos, solo requerirán ablandamiento aquellos de
consistencia más firme.
Métodos de hidratación
para materiales secos - Colocar la muestra en recipiente adecuado con (a) agua,
en cantidad de 20 a 30 veces el volumen de muestra. Luego colocar sobre una
plancha calefactora o una tela metálica, calentar suavemente hasta ebullición y
mantenerla durante 5 minutos o si no hierve, se procede a ablandarlo
hirviéndolo durante 5 minutos más en agua con detergente y ensayando su
consistencia; o (b) solución de hidratación, preparada con cinco partes de
agua, cuatro partes de etanol, una parte de glicerol y cinco gotas de
detergente comercial, para cada 200 mL de solución, en estufa a 60ºC, por un
período variable, de acuerdo con la textura del material. Las flores y las
hojas tienden a hidratarse en pocos minutos, mientras que los materiales duros,
como cáscaras y semillas exigen un tiempo variable en agua caliente (a), de
acuerdo a su grado de lignificación, u horas o días en la solución de
hidratación (b). En la hidratación directa en agua (a), cuidar el tiempo, ya
que puede ocurrir demasiado ablandamiento, impidiendo la observación al
microscopio óptico. En las dos técnicas, ensayar periódicamente la consistencia
del material. Para futuros análisis, determinar el tiempo que cada droga
vegetal necesita para adquirir una consistencia tal que permita su corte.
Obtención de los cortes
histológicos - Una vez hidratados y ablandados, proceder a la preparación de
los cortes de los órganos vegetales o sus partes. En general los cortes son
transversales al eje del órgano, y en algunas monografías se especifican cortes
longitudinales o tangenciales (cortezas, raíces etc.), o bien paradérmicos para
la observación de la epidermis de órganos foliáceos (hojas, sépalos y pétalos).
Los cortes a mano alzada se realizan con ayuda de cuchillas de corte. Las
estructuras más pequeñas o delgadas requieren que la muestra se fijen o
incluyan en material adecuado. Las secciones de mejor calidad se pueden obtener
con la ayuda de micrótomos. Seleccionar los cortes más delgados para
observación al microscopio a 10x.
Coloración y montaje de
láminas - Sumergir los cortes en solución de hipoclorito de sodio al 50 % para
eliminar el contenido celular. Dejar actuar hasta que los cortes se vuelvan
transparentes (no más de 10 a 15 minutos). Lavar los cortes con agua destilada
hasta la eliminación del hipoclorito de sodio, hasta pH neutro. Colocar los
cortes en solución de azul de toluidina al 0,05 %, durante 10 segundos. Lavar
con agua destilada, seguido de solución de ácido acético al 0,5 % y nuevamente
con agua destilada. Colocar entre porta y cubreobjetos con 2 a 3 gotas de una
mezcla de glicerina-agua destilada (1:1) y observar al microscopio óptico a 10x
y 40x. Las paredes celulósicas se tiñen de rosa púrpura. Las paredes lignificadas
y las paredes con taninos se tiñen de color azul verdoso brillante. [NOTA: la
coloración así obtenida no es estable.]
DISOCIACIÓN DE TEJIDOS
Este método se emplea
principalmente para el análisis de hojas, tallos herbáceos y cortezas. Los
cristales se mantienen intactos. Los granos de almidón pierden su estructura
característica.
Colocar en un vaso de
precipitados de 30 mL una porción del material vegetal. Agregar 10 mL de
solución de hidróxido de sodio al 5 % y llevar a ebullición durante 5 minutos.
Enfriar. Trasvasar a un tubo de centrífuga. Centrifugar durante 2 minutos.
Descartar la solución sobrenadante. Lavar con agua destilada. Colocar una
porción del centrifugado sobre un portaobjetos con 2 o 3 gotas de una mezcla de
glicerina-agua (1:1). Colocar el cubreobjetos y presionar. Observar al
microscopio óptico a 10x y 40x.
Observación de la droga en
polvo
Tomar 1 a 2 mg de la droga
en polvo y colocar una pequeña porción con un pincel fino y suave sobre un
portaobjetos. Agregar 2 o 3 gotas de solución de ácido láctico al 5 %
(diafanizante), y si es necesario, antes de colocar el cubreobjetos, adicionar
1 o 2 gotas de agua o de glicerol-etanol (1:1), mezclando bien con el pincel.
Colocar el cubreobjetos. Observar al microscopio óptico a 10x y 40x.
Determinación del índice
de estomas
El índice de estomas es
utilizado en el análisis de estructuras laminares, como hojas, folíolos y
brácteas, contando el número de estomas en una determinada área de la
epidermis. Para este recuento es necesario preparar porciones de alrededor de
0,5 cm por 0,5 cm de lámina foliar, sumergirlas en una mezcla de 10 mL de
hidrato de cloral y agua (5:2), en un vaso de precipitado, llevar a ebullición
por 10 a 15 minutos, hasta que el material se vuelva transparente. Realizar la
operación en campana de extracción. Colocar una porción de la hoja preparada en
un portaobjetos, con la epidermis abaxial hacia arriba. Para hojas muy gruesas
seccionar cada porción próxima a la epidermis inferior, cuidando que esta parte
sea colocada correctamente en el portaobjetos, con la capa epidérmica hacia el
cubre objetos. Agregar de 2 a 3 gotas de la mezcla de hidrato de cloral y agua
(5:2) y cubrir con un cubreobjetos. Observar al microscopio óptimo, en 10x.
Contar las células epidérmicas y los estomas que aparecen en el área. El índice
se calcula siguiendo la ecuación 100S/(E+S), siendo S el número de estomas en
un área determinada de la superficie de la hoja y E el número de células
epidérmicas, incluyendo los tricomas existentes en el mismo campo microscópico
observado. Para cada muestra, efectuar y calcular la media de diez
determinaciones, como mínimo.
Reacciones Histoquímicas
Las reacciones pueden ser
realizadas con material fresco o seco seccionado, material cortado en micrótomo
o polvo de la droga vegetal. El material se coloca adecuadamente distribuido en
un porta objeto, se agrega 1 o 2 gotas del reactivo. Colocar el cubreobjetos y
observar al microscopio óptimo a 10x y/o 40x.
Almidón. Agregar 1 o 2
gotas de Solución de Iodo SR diluida (1:5) en agua. Los granos de almidón se
colorean de azul o azul-violeta.
Concreciones de carbonato
de calcio (cistolitos) y de cristales de oxalato de calcio. Agregar 1 o 2 gotas
de ácido clorhídrico 2 M o ácido acético al 6% (p/v). La presencia de carbonato
de calcio está indicada por la formación de burbujas. Los cristales de oxalato
de calcio, demoran más tiempo en disolverse, no desprenden burbujas y son
insolubles en ácido acético al 6% (p/v).
Hidroxiantraquinonas.
Agregar una gota de hidróxido de potasio al 5% (p/v). Las células que contienen
1,8-dihidroxiantraquinonas se colorean de rojo.
Inulina. Agregar 1 gota de
solución de 1-Naftol al 20% en metanol, seguido de 1 gota de ácido sulfúrico.
Los esferocristales de inulina se colorean de rojo o marrón-rojizo y se
disuelven.
Lignina. Agregar 1 gota de
Floroglucina SR, calentar rápidamente la lámina y adicionar una gota de ácido
clorhídrico al 25% (p/v). La lignina se colorea de rojo.
Lípidos (incluyendo
cutina, ceras y suberina). Agregar 1 o 2 gotas de Reactivo de Sudan III SR o
Sudan IV SR dejando en contacto durante 10 minutos, lavar con etanol al 70%
(v/v). Lípidos, cutina y suberina se colorean de naranja rojizo a rojo luego de
un corto tiempo.
Pectinas y mucílagos.
Sumergir la muestra seca en Solución de Tionina, dejando reposar por 15
minutos, lavar con etanol al 20% (v/v). Los mucílagos aparecen como glóbulos
esféricos de color rojo-violeta, mientras que la celulosa, la pectina, y
tabiques lignificados se colorean de azul o azul-violeta. Los mucílagos también
aparecen como fragmentos esféricos dilatados y transparentes sobre un fondo
negro, con la adición de 1 gota de tinta nanquín sobre la muestra seca.
Proteínas. Realizar el
procedimiento solamente sobre material fresco. Agregar ninhidrina a 0,5% (p/v)
en etanol absoluto, mantener a 37°C por 24 horas. Lavar con etanol absoluto
seguido de agua destilada, agregar Reactivo de Schiff SR y dejar en contacto de
10 a 30 minutos. Lavar con agua y agregar bisulfito de sodio al 2% (p/v), dejar
en contacto de 1 a 2 minutos. Lavar con agua corriente durante 10 a 20 minutos.
Las proteínas se colorean de rojo-púrpura.
Saponinas. Agregar una
gota de ácido sulfúrico. Ocurre una secuencia de color amarilla, rojo y violeta
o azul-verdoso.
Taninos. Adicionar cloruro
férrico 5% (p/v) y una pequeña cantidad de carbonato de sodio, dejar en
contacto por 2 a 3 minutos, lavar con agua destilada. Los taninos se colorean
azul-verdoso oscuro.
DETERMINACIÓN DE MATERIA
EXTRAÑA
Materia extraña es
cualquier material que no esté comprendida en la definición de droga de la
monografía correspondiente. Las drogas deben estar libres de hongos, de
insectos y de otras contaminaciones de origen animal. Salvo que se indique lo
contrario, el porcentaje de elementos extraños no debe ser superior al 2% m/m.
La materia extraña de la droga se puede clasificar en tres tipos: (a) partes de
organismos u organismos que provienen de las drogas, exceptuando aquellos
incluídos en la definición y descripción de la droga, por encima del limite de
tolerancia especificado en la monografía; (b) cualquier organismo, partes o
productos de organismos no especificados en la definición y descripción de la
droga, en su respectiva monografía; y (c) impurezas de naturaleza mineral u
otras sustancias no relacionados con la droga. Durante el almacenamiento, los
productos deben mantenerse en un área limpia, de modo de evitar su
contaminación. Deben tomarse precauciones especiales para evitar la
proliferación de hongos dado que algunos de ellos pueden generar toxinas.
Procedimiento
A menos que se especifique
de otro modo en la monografía correspondiente, obtener por cuarteo las
siguientes cantidades de muestra:
- Raíces, rizomas,
cortezas, planta entera y partes aéreas: 500 g;
- Hojas, flores, frutos
y semillas: 250 g;
- Drogas vegetales en
fragmentos de 0,5 g o menores: 50 g;
- Polvos: 25 g
Extender la muestra en una
capa delgada y sobre una superficie plana. Separar manualmente la materia
extraña a la droga, inicialmente a ojo desnudo y, luego, con auxilio de una
lente de aumento (5 a 10X). Pesar el material separado y determinar el
porcentaje de materia extraña a partir de la cantidad de droga sometida al
ensayo.
DETERMINACIÓN DE AGUA EN
DROGAS VEGETALES
Para la determinación de
agua en drogas vegetales se emplean tres métodos: método gravimétrico
(desecación), método azeotrópico (destilación con tolueno) y método volumétrico
(Karl Fischer). El primero, técnicamente es el más simple y rápido, no es aplicable
cuando la droga contiene sustancias volátiles. Los demás métodos requieren
equipamientos especiales y comprenden técnicas más complejas.
Preparación de la muestra
Reducir la muestra por
corte, granulación o fragmentación de las drogas no pulverizadas o trituradas,
de forma de limitar la dimensión de sus componentes a aproximadamente 3 mm de
espesor. Las semillas o frutos de dimensiones inferiores a 3 mm se deben
fragmentar. Evitar el empleo de molinos de alta velocidad para preparar la
muestra y tomar las precauciones necesarias para no modificar el contenido de
humedad de la muestra.
Método gravimétrico
Transferir 1 a 10 g, o lo
especificado en la monografía, exactamente pesados de la muestra preparada
conforme a las instrucciones anteriores, en un pesa-filtro, exactamente pesado,
previamente desecado en las mismas condiciones a ser adoptadas para la muestra,
durante 30 minutos. Desecar la muestra entre 100 ºC y 105 ºC durante 5 horas,
hasta peso constante, es decir que la diferencia entre dos pesadas sucesivas no
superen el 0,25% del peso de la muestra. Calcular el porcentaje de agua en
relación a la droga seca.
DETERMINACIÓN DE CENIZAS
Las cenizas totales
incluyen cenizas fisiológicas y cenizas no-fisiológicas.
Determinación de cenizas
totales
Procedimiento
Pesar, analíticamente,
cerca de 3 g de muestra pulverizada, o la cantidad especificada en la
monografía, transferir a un crisol de porcelana previamente tarado. Distribuir
la muestra uniformemente en el crisol e incinerar aumentando, gradualmente, la
temperatura hasta, un máximo de 600 ± 50ºC, hasta que todo el carbono se haya
eliminado. Puede emplearse un gradiente de temperatura (30 minutos a 200ºC, 60
minutos a 400ºC y 90 minutos a 600ºC). Enfriar en desecador y pesar. En los
casos en que el carbono no pueda ser eliminado totalmente, enfriar el crisol y
humedecer el residuo con alrededor de 2 mL de agua o solución saturada de
nitrato de amonio. Evaporar hasta sequedad en baño de agua y, luego, colocarlo
sobre una placa caliente, e incinerar hasta que la diferencia entre dos pesadas
sucesivas no sea mayor que 1,0 mg. Calcular a porcentaje de cenizas en relación
a la droga seca.
Determinación de cenizas
sulfatadas
Procedimiento
Calentar un crisol de
porcelana al rojo durante 10 minutos, dejar enfriar en un desecador y pesar.
Pesar exactamente cerca de 1,0 g de la droga en un crisol previamente pesado y
humedecer la droga con ácido sulfúrico concentrado. Carbonizar en el quemador Bunsen.
Humedecer nuevamente con ácido sulfúrico concentrado, carbonizar e incinerar
con calentamiento gradual a 800ºC. Enfriar, pesar nuevamente e incinerar
durante 15 minutos más. Repetir este procedimiento hasta que la diferencia
entre dos pesadas sucesivas no sea mayor a 1,0 mg.
Determinación de cenizas
insolubles en ácido
Las cenizas insolubles en
ácido constituyen el residuo obtenido en calentamiento a ebullición de cenizas
totales o sulfatadas, con ácido clorhídrico diluido luego de la filtración, lavado
e incineración. El método se emplea para la determinación de sílice y
constituyentes silíceos de la droga.
Procedimiento
Calentar a ebullición las
cenizas obtenidas según se indica en Cenizas totales, con 25 ml de ácido
clorhídrico 2 M durante 5 minutos en un crisol cubierto por un vidrio de reloj.
Lavar el vidrio de reloj con 5 mL de agua caliente, juntando el agua de lavado
en un crisol. Recolectar el material insoluble en ácido en un papel de filtro
con tenor de ceniza conocida, lavado con agua caliente hasta que el filtrado se
vuelva neutro. Transferir el papel de filtro conteniendo el residuo al crisol
original, secar sobre una plancha caliente e incinerar a alrededor de 500ºC
hasta que la diferencia entre dos pesadas sucesivas no sea mayor a 1,0 mg.
Calcular el porcentaje de cenizas insolubles en ácido respecto de la droga
seca.
DETERMINACIÓN DE ACEITES
VOLÁTILES/ESENCIALES
El tenor de aceites
volátiles en drogas vegetales se lleva a cabo por el proceso de
hidrodestilación o arrastre con vapor, en un equipamiento descrito a
continuación.
El equipo (Figura X),
confeccionado en vidrio resistente, de calidad apropiada, comprende:

Figura X. Aparato para la
determinación del contenido de aceites volátiles en drogas vegetales mediante
el proceso de hidrodestilación
1) balón de fondo redondo
de 500 mL a 1000 mL de capacidad, de cuello corto, provisto de una junta 24/40,
hembra;
2) condensador, adaptable
al baño por medio de una junta esmerilada 24/40, macho, construido en pieza
única de vidrio, comprendiendo las partes descritas a seguir, con las
respectivas medidas:
2.1) tubo vertical (AC) de
240 mm de ancho y 13-15 mm de diámetro interno;
2.2) tubo doblado, con
hilos (CD) y (DE) cada uno de 150 mm de ancho y diámetro interno de 10 mm;
2.3) condensador de bolas,
tipo Allihn (FG), de 150 mm de ancho y diámetro interno de 15 mm (bolas) y 8-10
mm (en los puntos más estrechos);
2.4) tapón (junta
esmerilada 14/20) (K’) conteniendo un orificio de alrededor de 1 mm de
diámetro, que obtura una salida lateral (K) provista de una junta esmerilada
14/20 hembra, en la extremidad;
2.5) tubo (GH) de 30-40 mm
de ancho y 7-8 mm de diámetro interno, formando las partes (HK) un ángulo (GHK)
de 35º;
2.6) alargamiento en forma
de pera (J) de 3 mL de capacidad;
2.7) tubo (JL) provisto de
escala graduada de 100-110 mm; de 1 mL de capacidad y subdividida en 0,01 mL;
2.8) alargamiento en forma
de bola (L) de aproximadamente 2 mL de capacidad;
2.9) válvula de 3 vías;
2.10) tubo de conexión
(BM) de 7-8 mm de diámetro, provisto de un tubo de seguridad. El punto de
inserción (B) se encuentra a 20 mm por encima de la parte más alta de la escala
graduada;
3) dispositivo de
calentamiento apropiado que permite una regulación precisa;
4) soporte vertical con un
anillo horizontal cubierto con material aislante.
Utilizar un aparato
perfectamente limpio. Después de seco, debe ser montado en un lugar libre de
corrientes de aire. La escala graduada debe ser evaluada y si es necesario,
establecer un factor de corrección para cada aparato. Proceder a la valoración
según la naturaleza de la droga en ensayo.
Procedimiento
Llevar a cabo el ensayo de
acuerdo con la naturaleza de la droga a ser examinada. Transferir al balón el
volumen de líquido indicado en la monografía correspondiente, y fragmentos de
plato poroso o perlas de vidrio. Adaptar el condensador al balón. Introducir
agua a través del tubo de llenado (N) hasta que alcance el nivel B. Quitar el
tapón (K’) y transferir la cantidad indicada de xileno empleando una pipeta con
abertura (K). Colocar el tapón (K’) asegurándose que los orificios de (K) y
(K’) coincidan entre sí. Calentar el líquido en el balón hasta ebullición y
ajustar la velocidad de destilación a 2 a 3 mL por minuto, a menos que se
especifique de otro modo en la monografía correspondiente.
Para determinar la
velocidad de destilación, disminuir el nivel de agua por medio de la válvula de
tres vías hasta que el menisco se encuentre en la marca inferior a (ver Figura
X). Cerrar la válvula y medir el tiempo que toma el líquido en alcanzar la
marca superior b. Abrir la válvula y continuar con la extracción durante 30 minutos,
modificando el calentamiento para regular la velocidad de destilación. Dejar
enfriar y leer el volumen de xileno en el tubo graduado después de por lo menos
10 minutos.

Figura XX. Indicación para
la determinación de la velocidad de destilación.
Introducir en el balón la
cantidad de droga descripta en la monografía y destilar como se describe
arriba, por el tiempo y la velocidad indicada en la monografía. Finalizada la
operación, dejar enfriar por 10 minutos y leer el volumen de aceite volátil
recogido en el tubo graduado. Restar de la lectura el volumen de xileno
determinado anteriormente. La diferencia representa la cantidad de aceite
volátil contenido en la muestra. Calcular el resultado en mililitros de aceite
volátil cada 100 g de la droga.
Cuando la determinación
del aceite volátil se emplee para fines analíticos, la obtención de la mezcla
de aceite volátil y xileno libre de agua se realiza como se detalla a
continuación: quitar el tapón (K’) y transferir 1,1 mL de una solución de
fluoresceinato de sodio al 0,1 % y 0,5 mL de agua. Disminuir el volumen de la
mezcla de aceite volátil y xileno dentro del tubo L por medio de la válvula de
tres vías. Dejar en reposo durante 5 minutos y descargar la mezcla lentamente
hasta alcanzar justo el nivel de la válvula (M). Abrir la válvula en el sentido
contrario a las agujas del reloj de manera tal que el agua fluya fuera del tubo
de conexión (BM). Lavar el tubo, primero con acetona y luego con tolueno,
introducidos por el tubo de llenado (N). Girar la llave en el sentido contrario
a las agujas del reloj de manera tal que se pueda recuperar la mezcla de xileno
y aceite volátil en un recipiente apropiado.
DETERMINACIÓN DE ACEITES
FIJOS
La determinación de
aceites fijos se basa en la extracción con un solvente que, después de ser
evaporado, deja como residuo el aceite cuya cantidad es determinada por pesada.
En caso que la muestra
contenga una cantidad elevada de componentes hidrosolubles (carbohidratos,
urea, ácido láctico, entre otros), puede ser necesario un pretratamiento de la
muestra a fin de evitar la interferencia en la determinación de materias
grasas. Para ello, colocar la muestra pesada en un embudo conteniendo un papel
de filtro, lavar con agua y secar el residuo en estufa a 105 ºC durante 2
horas.
Emplear el aparato de
Soxhlet (Figura 1). El equipamiento, confeccionado en vidrio resistente, de
calidad apropiada, comprende un balón de fondo redondo (A), con 500 mL a 1000
mL de capacidad, conectado al extractor Soxhlet (B) y a un condensador de
reflujo (C).
Antes de la utilización,
el aparato debe estar adecuadamente limpio. Después de secado, debe ser montado
en un lugar protegido de corrientes de aire.

Procedimiento
Transferir, exactamente,
alrededor de 10 g de droga previamente desecada conforme a lo descrito en
Determinación de agua en drogas vegetales, Método gravimétrico, y transferir al
aparato extractor Soxhlet (B), cubriéndolo con un algodón desengrasado. Pesar
el balón (A) limpio y seco (conteniendo fragmentos de porcelana o perlas de
vidrio) y colocarlo sobre el aparato en baño de agua, tomando la precaución de
asegurar que la junta esmerilada del balón quede bien sellada (se recomienda
realizar la operación en campana de extracción). Transferir al extractor éter
de petróleo en cantidad suficiente para realizar tres vueltas de sifón y
colocar el condensador de reflujo (C). Proceder a la extracción con
calentamiento suficiente para mantener el solvente en ebullición moderada
durante 4 horas.
Concluida la extracción,
esperar que se enfríe, transferir el contenido del cartucho a un mortero de
porcelana y juntar una cantidad aproximadamente igual de arena lavada y seca.
Pulverizar la droga y transferir nuevamente, al interior del cartucho, y al
extractor. Reiniciar y mantener la extracción en las condiciones anteriores por
un período adicional de 2 horas. Separar el balón del aparato y evaporar el
solvente (de preferencia por destilación en corriente de dióxido de carbono).
Transferir el balón a una estufa a 105 ºC, enfriar y pesar. Repetir la
operación hasta obtener peso constante. Calcular el porcentaje de aceites fijos
en la droga en relación a la masa de droga pesada y la masa de aceite obtenido.
DETERMINACIÓN DEL ÍNDICE
DE ESPUMA
Pesar, exactamente, 1 g de
material vegetal reducido a polvo fino (malla de 180 µm) y transferir a un
erlenmeyer conteniendo 50 mL de agua hirviendo. Mantener a ebullición durante
30 minutos. Enfriar, filtrar en un matraz de 100 mL. Llevar a volumen, a través
del filtro, hasta 100 mL. Distribuir el filtrado obtenido (por decocción), en
10 tubos de ensayo con tapa (16 mm de diámetro por 16 cm de altura), en series
sucesivas de 1, 2, 3, hasta 10 mL, y ajustar a volumen de líquido en cada tubo
a 10 mL con agua. Tapar los tubos y agitarlos con movimientos verticales por 15
segundos, con dos agitaciones por segundo. Dejar en reposo por 15 minutos y
medir la altura de la espuma.
Si la altura de la espuma
de todos los tubos es inferior a 1 cm, el índice de espuma es menor que 100.
Si, en cualquiera de los tubos, la altura de la espuma medida es 1 cm, la
dilución del material vegetal en ese tubo (A) es el índice observado. Si ese tubo
es el primero o el segundo en la serie, es necesario hacer una dilución
intermedia, mediante el mismo método descrito anteriormente, para obtener un
resultado mas preciso. Si la altura de la espuma es mayor que 1 cm en todos los
tubos, el índice de espuma es mayor que 1000. En ese caso, la determinación
debe hacerse con una nueva serie de diluciones de la decocción para obtener un
resultado preciso. El índice de espuma es calculado según la ecuación 1000/A,
siendo A el volumen, en mililitros, de la decocción usada para la preparación
de la dilución en el tubo donde la espuma fue observada.
DETERMINACIÓN DE
SUSTANCIAS EXTRAÍBLES
Este método determina la
cantidad de constituyentes activos extraídos con solventes de una determinada
cantidad de material vegetal. Es empleado para materiales para los cuales aún
no existe un ensayo químico o biológico adecuado.
MÉTODO A: EXTRACCIÓN EN
FRÍO
Pesar un Erlenmeyer de 250
mL, con boca esmerilada y transferir en él, exactamente, alrededor de 4,0 g de
droga vegetal seca y pulverizada. Macerar, con 100 mL de solvente especificado
en el ensayo para la droga vegetal, durante 6 h, agitando frecuentemente, y
dejar en reposo por 18 h. Filtrar, rápidamente, tratando de no perder cantidad
de solvente; transferir 25 mL del filtrado a un cristalizador previamente
pesado y evaporar hasta sequedad en baño de agua. Secar en estufa a 105ºC hasta
peso constante. Calcular el porcentaje de material extraído en mg/g de material
vegetal seco.
MÉTODO B: EXTRACCIÓN EN
CALIENTE
Pesar un Erlenmeyer de 250
mL, con boca esmerilada, transferir a éste, exactamente, alrededor de 4,0 g de
droga vegetal seca y pulverizada. Agregar 100 mL del solvente especificado en
el ensayo para la droga vegetal y pesar para obtener el peso total, incluyendo el
frasco. Tapar, agitar bien y dejar descansar por 1 h. Montar sobre un
condensador de reflujo y calentar suavemente por 1 h, enfriar y pesar. Llevar a
peso original con el solvente utilizado. Agitar y filtrar, rápidamente, por
medio de un filtro seco. Transferir 25 mL del filtrado a un cristalizador
previamente tarado y evaporar hasta sequedad en baño de agua. Secar en estufa a
105ºC hasta peso constante. Calcular el porcentaje de material extraído en mg/g
de material vegetal seco.
MÉTODO C: EXTRACCIÓN POR
SOXHLET
Pesar, exactamente,
alrededor de 2 g de droga y transferir a un cartucho para extracción en
Soxhlet, previamente tarado y seco. Introducir en el balón para extracción 0,2
g de hidróxido de sodio y etanol absoluto en cantidad suficiente. Extraer por 5
horas, retirar el cartucho con el residuo y secarlo en estufa a 105ºC hasta
peso constante. Calcular el porcentaje de materiales extraídos en mg/g de
material vegetal seco. (Determinación de agua en drogas vegetales).
DETERMINACIÓN DEL ÍNDICE
DE AMARGOR
El índice de amargor de
una sustancia, un líquido o un extracto es la inversa de la dilución límite a
la cual aún se percibe sabor amargo. Está determinado por la comparación con
clorhidrato de quinina cuyo índice de amargor es de 200.000.
Determinación del factor
de corrección
Para este ensayo, se
recomienda que el grupo de experimentadores esté constituido por un mínimo de
seis personas. Cada experimentador debe enjuagar su boca con agua potable antes
del ensayo. Para corregir las diferencias individuales del sabor amargo entre
los experimentadores es necesario determinar un factor de corrección para cada
miembro.
Solución madre
Disolver 0,1 g de
Clorhidrato de quinina R en agua potable y diluir a 100 mL con el mismo
solvente. Diluir 1,0 mL de esta solución a 100 mL con agua potable.
Soluciones de referencia
Preparar una serie de
diluciones colocando 3,6 mL de la solución madre en el primer tubo, y aumentar
el volumen en 0,2 mL en cada tubo secuencialmente hasta un volumen de 5,8 mL.
Llevar el volumen total de todos los tubos a 10,0 mL con agua potable.
Determinar la mayor
dilución a la cual se observa aún el sabor amargo. Colocar en la boca 10 mL de
la solución más diluida y pasarla de un lado a otro por debajo de la lengua
durante 30 segundos. Si no se detecta un sabor amargo nítido, desechar la
solución y esperar un minuto. Enjuagar la boca con agua potable. Después de 10
minutos, probar la solución siguiente en orden creciente de concentración.
Calcular el factor de corrección (k) para cada experimentador usando la expresión
siguiente:
n = volumen en mililitros,
de la mayor dilución de la solución madre en la que se detectó el sabor amargo
nítido.
Aquellos experimentadores
que no detectan sabor amargo nítido en la solución preparada con 5,8 mL de la
solución madre, deben ser excluidos del ensayo.
Preparación de la muestra
Si es necesario, reducir
la muestra a polvo (710). Pesar 1,0 g de la muestra y agregar 100 mL de agua
potable hirviendo. Calentar en baño-maría por 30 minutos, agitando
continuamente. Dejar enfriar y compensar el volumen de agua evaporada con agua
potable. Agitar vigorosamente y filtrar, descartando los primeros 2 mL del
filtrado. El filtrado se rotula como C-1 y tiene un factor de dilución (FD) de
100.
Para muestras líquidas,
tomar 1 mL y diluir con un solvente apropiado hasta 100 mL y rotularlo como
C-1.
Determinación del índice
de amargor
Soluciones muestra:
10,0 mL de C-1 se diluyen
con agua a 100 mL: C-2 (FD 1000)
10,0 mL de C-2 se diluyen
con agua a 100 mL: C-3 (FD 10 000)
20,0 mL de C-3 se diluyen
con agua a 100 mL: C-3A (FD 50 000)
10,0 mL de C-3 se diluyen
con agua a 100 mL: C-4 (FD 100 000)
Comenzando por la dilución
C-4, cada experimentador debe definir la dilución a la cual percibe el sabor
amargo nítido. Esta solución se rotula como D. La FD de esta solución
corresponde a Y.
Se prepara la siguiente
secuencia de diluciones a partir de la solución D:
|
Solución D (mL)
|
1,2
|
1,5
|
2,0
|
3,0
|
6,0
|
8,0
|
|
Agua potable (mL)
|
8,8
|
8,5
|
8,0
|
7,0
|
4,0
|
2,0
|
Determinar el volumen en
mililitros de la solución D, en la cual aún diluida a 10 mL se percibe el sabor
amargo nítido (X).
Calcular el Índice de
Amargor para cada experimentador conforme a la fórmula:
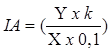
El Índice de Amargor de la
muestra es el valor de la media de los experimentadores.
DETERMINACIÓN DEL ÍNDICE
DE HINCHAMIENTO
El índice de hinchamiento
es la medida del volumen ocupado por el hinchamiento de 1 g de la droga,
mediante la adición de agua u otro agente turgente, bajo condiciones definidas.
Realizar, simultáneamente,
como mínimo, tres determinaciones. Pesar, exactamente, 1 g de la droga vegetal
pulverizada y colocar en una probeta de 25 mL con boca esmerilada. El ancho de
la parte graduada debe ser de, aproximadamente, 125 mm y el diámetro, interno,
próximo a 16 mm, subdividido en 0,2 mL, marcado de 0 a 25 mL, de forma
ascendente. Agregar 25 mL de agua, u otro agente definido, y agitar cada 10
minutos, por una hora. Dejar la mezcla en reposo por 3 horas, a temperatura
ambiente. Medir el volumen en mililitros ocupados por el material de la planta
más el mucílago o cualquier otro material adherido restando del volumen inicial
de la droga. Calcular el valor medio obtenido a partir de varias
determinaciones individuales realizadas y referirlo a 1 g de material vegetal.
REACTIVOS
Floroglucina SR: Disolver
1 g de floroglucinol en etanol y diluir en 100 mL con el mismo solvente,
conservando en recipiente bien cerrado y al abrigo de la luz.
Iodo SR (Solución acuosa
de iodo – iodurada): Disolver 1 g de iodo en 100 mL de agua, agregar 2 g de
iodato de potasio, agitar, dejar en reposo por algunas horas y filtrar en lana
de vidrio. Conservar en frasco color ámbar bien cerrado.
1-Naftol SR: Disolver 20 g
de 1-naftol en 100 mL de etanol R. Preparar para uso inmediato.
Reactivo de Schiff SR (=
fucsina decolorada): Disolver 1 g de fucsina básica en 600 mL de agua,
adicionar 100 mL de sulfito de sodio anhidro a 10% (p/v). Enfriar externamente
con hielo, con agitación. Agregar, lentamente, 10 mL de ácido clorhídrico,
diluir con agua en 1000 mL y filtrar. Si la solución oscurece, agitar con 0,2 a
0,3 g de carbón activado hasta decoloración, filtrando inmediatamente. Si aún
permanece la coloración rosácea, agregar de 2 a 3 mL de ácido clorhídrico y
agitar. Dejar en reposo durante 1 hora antes de la utilización, y mantener al
abrigo de la luz.
Solución de Sudan III:
Disolver 0,5 g de Sudan III en 100 mL de etanol 80%, calentando a 60°, enfriar
y filtrar.
Solución de Sudan IV:
Disolver 2,0 g de Sudan IV en 100 mL de etanol 92%, calentando a 60°, enfriar,
filtrar y agregar 5 mL de glicerina.
Solución de Tionina:
Preparar una solución de acetato de tionina al 0,2% en etanol 25 %. Sumergir la
muestra seca en la solución. Luego de 15 minutos lavar el exceso de reactivo
con 25% de etanol.
RESIDUO DE
AGROQUÍMICOS/AGROTÓXICOS
A los efectos de la
Farmacopea, un agroquímico/agrotóxico es cualquier sustancia o mezcla de
sustancias destinadas a prevenir, destruir o controlar cualquier plaga, las
especies no deseadas de plantas o animales que causan perjuicio o que
interfieren de cualquier otra forma en la producción, elaboración,
almacenamiento, transporte o comercialización de drogas vegetales. La
definición abarca además sustancias empleadas como reguladores de crecimiento,
hormonas, desfoliantes y desecantes así como cualquier otra sustancia aplicada
a los cultivos antes o después de la cosecha para prevenir su deterioro durante
el almacenamiento y transporte. Los residuos de agrotóxicos pueden estar
presentes en drogas vegetales y sus preparados y deben analizarse para
determinar su presencia
Límites
La presencia de
agroquímicos/agrotóxicos incluidos en el Convenio de Estocolmo y
agroquímicos/agrotóxicos no autorizados / no registrados por la legislación
vigente, no debe ser mayor a 0,03mg/kg de droga, aceptándose su presencia sólo
como producto de contaminación ambiental.
Para todos aquellos
agroquímicos/agrotóxicos encontrados en plantas medicinales, el Límite
Aceptable de Residuos (LARs) aceptado estará dado por la fórmula:

LARHD Límite aceptable de
Agroquímicos en mg/kg de droga
IDA Ingesta Diaria
Admisible (mg/kg de peso corporal) según FAO o la legislación vigente.
M peso corporal (kg) (se
toma como convención 60kg)
DDD Dosis diaria de droga
(kg)
El factor de 100
corresponde a la participación de la ingesta de la droga en la dieta diaria
(1%). Para casos especiales, este factor puede ser variado, según los hábitos
alimenticios de la población, debidamente documentados.
Los límites máximos de
agroquímicos/agrotóxicos en preparaciones herbarias están dados por la fórmula:
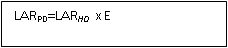
Donde
LARPD Límite aceptable de
residuos de agroquímicos/agrotóxicos en mg/kg del preparado de droga vegetal.
E= Factor de extracción
del agroquímico/agrotóxico, determinado experimentalmente. Es una medida de
la transferencia del agroquímico/agrotóxico de la droga vegetal a la
preparación farmacéutica.
Muestreo
Debe ser realizado de
acuerdo a los criterios establecidos en métodos generales.
Análisis cualitativo y
cuantitativo de residuos de agroquímicos/agrotóxicos
Los procedimientos
analíticos empleados deben ser validados de acuerdo al documento SANCO en su
versión más actualizada “Guidance Document on analytical quality control and
validation procedures for pesticide residues in food and feed” y satisfacer
mínimamente los siguientes criterios:
El método de extracción
elegido debe ser apropiado para la combinación de agroquímicos/agrotóxicos que
se pretende investigar y no provocar interferencias.
Se debe considerar las
interferencias posibles de la matriz, por ejemplo, interferencias de
compuestos azufrados en brasicáceas y aliáceas en la determinación de
ditiocarbamatos como CS2.
Las soluciones estándar y
soluciones muestra deben estar en el rango lineal del detector.
Los límites de detección y
cuantificación deben determinarse para cada combinación de agroquímicos/agrotóxicos
y matrices a ser analizadas.
La recuperación debe estar
entre el 70 y 110 %.
La repetitividad y
reproducibilidad del método no debe ser menor que la indicada en la Tabla 1.
Tabla 1.
|
Concentración de
pesticida (mg/kg)
|
Repetitividad
(±mg/kg)
|
Reproducibilidad
(±mg/kg)
|
|
0,01
|
0,005
|
0,01
|
|
0,1
|
0,025
|
0,05
|
|
1
|
0,125
|
0,25
|