Disposición
6766/2016
Guía para la
solicitud de Bioexenciones de Ingredientes Farmacéuticos Activos con
Requerimiento de Bioequivalencia
Bs. As., 29/06/2016
VISTO la Ley N° 16.463,
sus Decretos Reglamentarios N° 9763/64 y .° 150/92 (T.O. Decreto 177/93), la
Resolución ex Secretaría de Políticas, Regulación y Relaciones Sanitarias N.°
46/03, las Disposiciones ANMAT N° 5904/96, N° 3185/99 con sus complementarias y
modificatorias, N° 3311/01, N° 2807/02, N° 2819/04, N° 5040/06, N° 1746/07, N°
2446/07, N° 556/09, N° 758/09, N° 5743/09, N° 6677/10, N° 3113/10, N° 1263/12,
N° 4132/12, N° 4326/12, N° 4788/12, N° 1918/13 y N° 2434/13, el Informe de la
Organización Mundial de la Salud N° 992/2015 (Anexo 7), y el Expediente N°
1-47-1110-311-14-0 de la Administración Nacional de Medicamentos, Alimentos y
Tecnología Médica; y
CONSIDERANDO:
Que los estudios de
bioequivalencia en humanos han sido aceptados en los últimos 25 años como un
requisito sobre el cual se basan las agencias regulatorias de medicamentos para
establecer la equivalencia terapéutica de los productos farmacéuticos
multifuentes con respecto a un producto de referencia.
Que por Disposición ANMAT
N° 3185/99 se aprobaron las recomendaciones técnicas para la realización de
estudios de equivalencia contenidas en el documento denominado: “Cronograma
para exigencia de estudios de equivalencia entre medicamentos de riesgo
sanitario significativo”, y mediante disposiciones posteriores, esta
Administración Nacional ha ido incorporando ingredientes farmacéuticos activos
a la obligatoriedad de demostración de bioequivalencia.
Que la Organización
Mundial de la Salud (OMS) en su Serie de Informes Técnicos N° 937 —Anexo 8— ha
considerado al Sistema de Clasificación Biofarmacéutica (SCB) como una
herramienta válida de clasificación de aquellos principios activos que solo
requieren demostraciones de equivalencia in vitro exceptuándolos de estudios in
vivo.
Que el SCB se basa en la
solubilidad acuosa y la permeabilidad intestinal del principio activo; cuando
estas dos propiedades se combinan con la disolución del medicamento se obtienen
los tres factores que determinan la velocidad y la cantidad de principio activo
absorbido desde una forma farmacéutica oral de liberación inmediata,
permitiendo establecer la inferencia de la Bioequivalencia.
Que de acuerdo a los
principios del SCB surgen dos conceptos aplicables a los productos
farmacéuticos multifuentes: el de estudios de equivalencia in vitro y el de
bioexenciones.
Que la introducción del
SCB ha provocado un gran impacto en la práctica regulatoria al establecer las
pautas para la demostración de la bioequivalencia entre medicamentos mediante
ensayos de disolución in-vitro para las formas farmacéuticas sólidas orales de
liberación inmediata, limitando así los requerimientos de estudios in vivo.
Que las agencias de
medicamentos de los Estados Unidos (Food and Drug Administration, FDA) y de la
Comunidad Económica Europea (European Medicines Agency, EMA) han adoptado los
criterios del SCB y ambas coinciden en que la bioexención es justificada cuando
las drogas tienen alta solubilidad y alta permeabilidad (Clase I), son de
amplio margen terapéutico y el producto cumple con determinados criterios de
disolución.
Que la OMS en el documento
antes mencionado considera exceptuables de la demostración de equivalencia in
vivo a las drogas de Clase I y también extiende la bioexención a los productos
medicinales que contengan drogas de Clase III (baja permeabilidad / alta
solubilidad), criterio recientemente adoptado por la EMA.
Que la demostración de la
bioequivalencia mediante ensayos de disolución, permite reducir en forma
considerable los tiempos y costos de la realización de estudios in vivo,
reemplazar en algunos casos los estudios en humanos y agilizar la ejecución de
las políticas sanitarias vigentes con la finalidad de poner al alcance de la
población un número cada vez mayor de productos medicinales con equivalencia
establecida.
Que es de interés nacional
promover la adopción de lineamientos técnicos para mantener el nivel de
fiscalización dentro de los parámetros internacionales.
Que en la Disposición
ANMAT N° 758/09 se establecieron los requisitos necesarios para reemplazar los
estudios de bioequivalencia in vivo por estudios de disolución basados en el
Sistema de Clasificación Biofarmacéutico del IFA.
Que es necesario
establecer requisitos para la solicitud de Bioexención, en los casos de:
solicitudes de exención de los estudios de biodisponibilidad (BD) y/o
bioequivalencia (BE) in vivo para formas farmacéuticas sólidas orales de
liberación inmediata (FFSO-LI) en base al Sistema de Clasificación
Biofarmacéutico (SCB) y para formulaciones sólidas orales proporcionalmente
similares a otro producto cuya equivalencia haya sido demostrada mediante un
estudio in vivo o in vitro, y para cambios posteriores en formulaciones que
hayan demostrado BE.
Que a tales fines es
necesario adoptar una Guía que aporte las recomendaciones para su
implementación.
Que la aplicación de las
normas aludidas tiene como finalidad última la protección de la salud de la
población mediante la adopción de un modelo fiscalizador de gestión que destina
sus esfuerzos a la verificación continua de la eficacia, seguridad y calidad de
los productos que aquélla consume.
Que el Instituto Nacional
de Medicamentos y la Dirección General de Asuntos Jurídicos han tomado la
intervención de su competencia.
Que se actúa en virtud de
las facultades conferidas por el Decreto N° 1490 de fecha 20 de agosto de 1992
y el Decreto N° 101 del 16 de diciembre de 2015.
Por ello,
EL ADMINISTRADOR NACIONAL
DE LA ADMINISTRACIÓN
NACIONAL
DE MEDICAMENTOS, ALIMENTOS
Y TECNOLOGÍA MÉDICA
DISPONE:
ARTÍCULO 1° — Las
solicitudes de Bioexenciones de Ingredientes Farmacéuticos Activos con
Requerimiento de Bioequivalencia deberán efectuarse de acuerdo a la “Guía para
la solicitud de Bioexenciones de Ingredientes Farmacéuticos Activos con
Requerimiento de Bioequivalencia” que como Anexo forma parte integrante de la
presente disposición.
ARTÍCULO 2° — La presente
disposición entrará en vigencia a partir del día siguiente a su publicación en
el Boletín Oficial.
ARTÍCULO 3° — Regístrese.
Dése a la Dirección Nacional del Registro Oficial para su publicación.
Notifíquese a las Cámaras de Especialidades Medicinales (CILFA, CAEME,
COOPERALA, CAPGEN, CAPEMVEL, SAFYBI), Confederación Médica de la República Argentina
(COMRA) y la Confederación Farmacéutica Argentina (COFA). Cumplido, archívese.
— Dr. CARLOS CHIALE, Administrador Nacional, A.N.M.A.T.
ANEXO
Guía para la solicitud de
Bioexenciones de Ingredientes Farmacéuticos Activos con Requerimiento de
Bioequivalencia
1. INTRODUCCIÓN
Esta guía aporta
recomendaciones para las solicitudes de exención de los estudios de
biodisponibilidad (BD) y/o bioequivalencia (BE) in vivo para formas
farmacéuticas sólidas orales de liberación inmediata (FFSO-LI) en base al
Sistema de Clasificación Biofarmacéutico (SCB)*; para formulaciones sólidas
orales proporcionalmente similares a otro producto cuya equivalencia haya sido
demostrada mediante un estudio in vivo o in vitro; y para cambios posteriores
en formulaciones que hayan demostrado BE.
La presente guía describe:
a) brevemente los
fundamentos del SCB.
b) cuándo y cómo se pueden
solicitar bioexenciones para FFSO-LI.
c) los métodos
recomendados para determinar la solubilidad y la permeabilidad de los
ingredientes farmacéuticos activos (IFAS en adelante), y la cinética de
liberación-disolución in vitro de los productos farmacéuticos de liberación
inmediata.
d) la forma de tramitar
una solicitud de bioexención y los antecedentes que deben presentarse a la
Administración Nacional de Medicamentos Alimentos y Tecnología Médica (ANMAT).
* El SCB considera
Productos de Liberación inmediata a aquellos que presentan Patrones de rápida y
muy rápida disolución.
2. EL SISTEMA DE
CLASIFICACIÓN BIOFARMACÉUTICO
2.1 Descripción y
Fundamento Científico:
El SCB es un marco
científico para clasificar los ingredientes farmacéuticos activos (IFAS) en
base a su solubilidad acuosa y su permeabilidad intestinal. (1-6)
Cuando se combina con la
disolución y el análisis crítico de los excipientes del producto farmacéutico
oral de liberación inmediata, el SCB toma en cuenta los factores principales
que gobiernan la velocidad y extensión de la absorción del IFA desde el
producto farmacéutico: la composición de excipientes, la disolución,
solubilidad y la permeabilidad intestinal. (7-8)
Según el SCB, los IFAS se
clasifican de acuerdo a la siguiente tabla.
Tabla 1: Clasificación de
los IFAS en base al Sistema de Clasificación Biofarmancéutico.
|
Clase
|
Solubilidad
|
Permeabilidad
|
|
I
|
Alta
|
Alta
|
|
II
|
Baja
|
Alta
|
|
III
|
Alta
|
Baja
|
|
IV
|
Baja
|
Baja
|
2.1.1 Solubilidad
Un IFA se considera
altamente soluble cuando la dosis más elevada del producto farmacéutico para la
vía oral es soluble en un volumen de 250 ml o menos (dosis / solubilidad = 250
ml) en un medio acuoso y en un intervalo de pH comprendido entre 1,2 y 6,8 a 37
± 1°C. Por ejemplo, si un IFA se encuentra en forma de comprimidos de 500 mg,
850 mg y 1000 mg, ese IFA se considera altamente soluble cuando la dosis más
elevada (1000 mg) se disuelve en las condiciones anteriormente citadas. (9-10)
2.1.2 Permeabilidad
Se considera que un IFA es
altamente permeable cuando se establece que la fracción absorbida de la dosis
administrada en seres humanos es del 85% o más, basándose en una determinación
de balance de masa o en la comparación con una dosis de referencia intravenosa.
(11)
2.1.3 Cinética de
disolución de las FFSO-LI
A los fines de una
solicitud de bioexención, se considera que un producto farmacéutico es de muy
rápida disolución cuando 85% o más de la cantidad declarada se disuelve dentro
de los 15 minutos en un medio de disolución de pH 1,2; 4,5 y 6,8 a 37°C usando
el aparato II —paleta— de la Farmacopea Argentina, 7° Ed. Vol 1 (Aparato
II.FA.7ma) a 75 rpm o alternativamente el aparato I —canastillo— de la
Farmacopea Argentina, 7° Ed. Vol 1 (Aparato I.FA.7ma) a 100 rpm.
Se considera que un
producto farmacéutico es de rápida disolución cuando más del 85% de la cantidad
declarada se disuelve dentro de los 30 minutos en las mismas condiciones
mencionadas anteriormente.
Podrá solicitarse la
bioexención para productos farmacéuticos conteniendo IFAS de Clase I si los dos
productos a comparar, referencia y multifuente, pueden ser categorizados como
de disolución muy rápida o, cuando siendo de disolución rápida sus perfiles de
disolución resultan esencialmente similares.
Para productos
farmacéuticos conteniendo IFAS de Clase III, en cambio, la bioexención sólo
podrá ser solicitada si los dos productos a comparar, referencia y multifuente,
pueden ser categorizados como de disolución muy rápida.
2.2 Antecedentes
internacionales de las Bioexenciones:
Agencias Regulatorias
La tabla 2 muestra las
diferencias, actualmente existentes, entre las clases bioexceptuables para las
distintas agencias regulatorias y organizaciones de salud.
Tabla 2: Clases de IFAS
bioexceptuables, según diferentes agencias regulatorias y organizaciones de
salud.
|
Clase I
|
Clase II
|
Clase III
|
Clase IV
|
|
EMA
|
Si
|
-
|
Si
|
-
|
|
FDA
|
Si
|
-
|
-
|
-
|
|
OMS
|
Si
|
-
|
Si
|
-
|
|
ANMAT
|
Si
|
-
|
Si
|
-
|
(EMA: Agencia Europea de
Medicamentos; FDA: Agencia de Medicamentos de Estados Unidos de América; OMS:
Organización Mundial de la Salud)
2.3 Ámbito de aplicación
de las Bioexenciones en Argentina:
El método del SCB descrito
en esta guía puede emplearse solamente para justificar las bioexenciones de los
productos farmacéuticos fabricados conforme a las Buenas Prácticas de
Fabricación y Control de Medicamentos.
La aplicación de estas
exenciones se ha considerado para:
a) Formas farmacéuticas
sólidas orales de liberación inmediata que contienen IFAS pertenecientes a la
clase I y/o III.
b) Formulaciones
proporcionalmente similares a otro producto cuya equivalencia haya sido
demostrada mediante un estudio in vivo o in vitro.
c) Cambios posteriores a
la aprobación del registro de un producto farmacéutico que ha demostrado BE.
3. APLICACIÓN DE
BIOEXENCIONES
3.1 Bioexenciones basadas
en el SCB
3.1.1 Metodología para
clasificar los IFAS en base a su solubilidad, permeabilidad y determinación de
las características cinéticas de disolución de los productos multifuente.
3.1.1.1 Clasificación en
base a la solubilidad de los IFAS
Uno de los objetivos del
SCB es determinar la solubilidad en equilibrio de un IFA bajo condiciones
fisiológicas de pH, para lo cual se deberá determinar la solubilidad en función
del pH del IFA en estudio, en un medio acuoso a 37±1°C en tres pH comprendidos
entre 1,2-6,8 durante 24 horas. Además, se deberá evaluar un número suficiente
de condiciones de pH para definir con precisión el perfil de solubilidad versus
pH.
Para la determinación de
la solubilidad, resulta conveniente seleccionar las condiciones de pH
considerando las características de ionización del IFA en estudio. Por ejemplo,
cuando su pKa se encuentra entre 3-5, determinar la solubilidad a pH = pKa; pH
= pKa+1; pH = pKa-1, y a pH = 1,2 y 6,8. Se recomienda un mínimo de tres
mediciones de solubilidad en cada condición de pH debiendo ser el CV% menor que
5% (CV% = [desvío estándar / media aritmética]*100). Según la variabilidad del
estudio, es posible que sea necesario un mayor número de determinaciones para
proveer un cálculo de solubilidad confiable. Si el número de muestras es mayor
que 3, todas las réplicas deben ser consideradas en el cálculo del desvío
estándar.
Para ser utilizadas en los
estudios de solubilidad, se consideran apropiadas las soluciones reguladoras
estándares descritas en la Farmacopea Argentina Ed. 7° o en el capítulo
Reactivos, Indicadores y Soluciones de la Farmacopea de Estados Unidos.(12-13)
Si estas soluciones no resultaran apropiadas por razones físicas o químicas, se
tendrán que utilizar otras soluciones reguladoras justificando su uso. Se
deberá verificar el pH de la solución antes y después de agregar el IFA a la
solución reguladora.
Utilizar el método
tradicional de agitación en matraz u otros métodos con justificación que avale
la capacidad de los mismos de medir la solubilidad en equilibrio del IFA en
estudio.
Se deberá determinar la
concentración del IFA en las soluciones reguladoras seleccionadas, en las condiciones
de pH señaladas, empleando un ensayo validado que indique la estabilidad y que
pueda diferenciar el IFA de sus productos de degradación si los hubiere (p.
ej., cuantificación por HPLC). (14) Si se observa la degradación del IFA en
función de la composición de la solución reguladora y/o del pH, se deberá
comunicar el hecho junto con los demás datos de estabilidad recomendados en
esta guía. La estabilidad del IFA se evaluará en todas las condiciones
experimentales considerando la duración total del estudio (comparación con el
valor nominal, en un mínimo de tres repeticiones).
Complementariamente, se
deberá establecer la clasificación de solubilidad, calculando el volumen de un
medio acuoso suficiente para disolver la mayor concentración posológica de la
forma farmacéutica en la gama de pH de 1,2-6,8. De esta forma, se clasificará
un IFA como altamente soluble cuando la mayor dosis sea soluble en un volumen =
250 mL de medio acuoso en toda la gama de pH comprendida entre 1,2-6,8. (15-16)
3.1.1.2 Clasificación en
base a la permeabilidad de los IFAS
Se puede establecer o
referenciar la clasificación de la permeabilidad de un IFA en sujetos humanos
empleando los métodos de balance de masa, de BD absoluta o de perfusión
intestinal.
Los métodos recomendados
que no involucran a sujetos humanos, incluyen la perfusión intestinal in vivo o
in situ en un modelo animal apropiado (p. ej., ratas) y/o métodos de
permeabilidad in vitro, empleando tejidos intestinales extirpados o monocapas
de células epiteliales apropiadas (p. ej., monocapas de células Caco- 2).
Podrá ser suficiente un
solo método cuando la BD absoluta es del 85% o más, o cuando se recupera en la
orina el 85% o más, del IFA administrado.
Si un sólo método no
demuestra en forma concluyente la clasificación de permeabilidad, se deben
emplear dos métodos diferentes.
La estructura química y/o
ciertos atributos fisicoquímicos de un IFA (p. ej., el coeficiente de partición
en sistemas apropiados) pueden proveer información preliminar útil acerca de
sus características de permeabilidad (método in sílico). (3) En ciertos casos,
los laboratorios pueden considerar el uso de tal información como antecedente
adicional de una clasificación determinada por estudios más robustos como los
realizados en humanos.
3.1.1.2.1 Estudios
farmacocinéticos en el hombre
• Estudios de balance de
masa
Para documentar la medida
de absorción de un IFA, es posible usar estudios farmacocinéticos de balance de
masa que utilizan isótopos estables o un IFA marcado radioactivamente. Sin
embargo, debido a que este método puede proveer cálculos altamente variables de
la absorción para muchos IFAS, no representa un método de elección,
recomendándose emplear otros métodos.
• Estudios de
biodisponibilidad absoluta
Se puede utilizar la
determinación de la BD oral usando la administración intravenosa como
referencia. Según la variabilidad de los estudios, se deberá incluir un número
suficiente de sujetos para proveer un cálculo confiable de la tasa de
absorción. Cuando se muestra que la BD absoluta de un IFA es del 85% o más, no
hacen falta datos adicionales para documentar la estabilidad del IFA en el
fluido gastrointestinal.
3.1.1.2.2 Métodos de
permeabilidad intestinal
Se pueden utilizar los
siguientes métodos para determinar la permeabilidad de un IFA en el sistema
gastrointestinal:
(1) estudios de perfusión
intestinal in vivo en seres humanos;
(2) estudios de perfusión
intestinal in vivo o in situ en modelos animales apropiados;
(3) estudios de
permeabilidad in vitro en tejidos intestinales extirpados humanos o animales;
(4) estudios de
permeabilidad in vitro en monocapas de células epiteliales.
Para IFAS transportados
pasivamente resultan adecuados los modelos animales in vivo o in situ, y los
métodos in vitro como los que usan monocapas de células epiteliales animales o
células humanas cultivadas.
La baja permeabilidad de
algunos IFAS observada en el hombre podría ser resultado de la expulsión de los
IFAS por transportadores de membranas como la glicoproteína de eflujo-P (P-gp).
Cuando dichos transportadores están ausentes en los modelos, o su grado de
expresión es bajo en comparación con el del hombre, puede haber una mayor
probabilidad de error en la clasificación de permeabilidad para un IFA sustrato
de los transportadores, en comparación con un IFA transportado en forma pasiva.
Por lo tanto, deberá caracterizarse la expresión de transportadores conocidos
en los modelos seleccionados. Se puede demostrar la expresión funcional de los
sistemas de transporte (p. ej., P-gp), empleando IFAS modelos seleccionados o
sustancias químicas sustrato de éstos en concentraciones que no saturen el
sistema de transporte (p. ej., ciclosporina A, vinblastina, rodamina 123),
mediante técnicas como estudios de transporte bidireccional, que muestran una mayor
velocidad de transporte en el sentido basolateral-apical en comparación con el
sentido apical-basolateral.
Esta guía recomienda
limitar el uso de métodos de prueba de permeabilidad no humanos para los IFAS
transportados por mecanismos pasivos. Para aplicar el SCB, se puede suponer un
transporte pasivo aparente, cuando se satisface una de las siguientes
condiciones:
• En el hombre se
demuestra una relación lineal (farmacocinética) entre la dosis (p. ej., en una
variedad posológica clínica relevante) y mediciones de área bajo la curva (área
debajo de la curva de concentración versus tiempo).
• En un modelo animal se
demuestra la falta de dependencia de la permeabilidad respecto de la
concentración inicial del IFA (p. ej., 0,01; 0,1 y 1 vez la mayor concentración
posológica disuelta en 250 mL), medida en el fluido de perfusión in vivo o in
situ.
• En un método de cultivo
de células in vitro apropiado, cuya expresión de proteínas transportadoras es
conocido (p. ej., P-gp), se demuestra la falta de dependencia de la
permeabilidad medida in vitro, respecto de la concentración inicial del IFA (p.
ej., 0,01; 0,1 y 1 vez la mayor concentración posológica disuelta en 250 mL) en
el fluido donante y respecto del sentido de transporte (p. ej., no existe una
diferencia estadísticamente significativa entre la velocidad de transporte
cuando se mide en el sentido apical-basolateral y cuando se mide en el sentido
basolateral-apical, para las concentraciones del IFA estudiadas).
Para demostrar la aptitud
de un método de permeabilidad determinado previsto para la aplicación del SCB,
se deberá establecer una relación categoría-orden entre los valores de
permeabilidad y los datos de absorción del IFA en sujetos humanos, empleando un
número suficiente de IFAS-modelo. Se recomiendan 3 IFAS-modelo para los
estudios de perfusión intestinal in vivo en el hombre y 15 IFAS modelo, para
los estudios de perfusión intestinal in vivo o in situ en animales y para
métodos de cultivo de células in vitro. Según la variabilidad del estudio, se deberá
usar un número estadísticamente apropiado de sujetos, animales, muestras de
tejidos extirpados o monocapas de células, para proporcionar un cálculo
confiable de la permeabilidad del IFA. La relación categoría-orden debe
permitir la diferenciación entre los IFA con atributos de permeabilidad
intestinal Baja, Moderada y Alta. (7) La adecuabilidad del método para la
determinación de la permeabilidad, podrá demostrarse mediante la utilización de
IFAS modelo seleccionados a partir de su tasa de absorción baja (< 50%),
moderada (50 - 85%) y alta (= 85%). En cada caso deberá existir un número
equitativo de IFAS entre las tres categorías. Los patrocinadores podrán
seleccionar compuestos de la lista de IFAS y/o sustancias químicas para los
cuales exista información disponible respecto del mecanismo de absorción, así
como también datos confiables de la cantidad de la absorción del IFA en el
hombre.
Después de demostrar la
aptitud de un método y mantener el mismo protocolo de estudio, no es necesario
ensayar nuevamente todos los IFAS modelo seleccionados para los estudios
posteriores que tengan como propósito clasificar un IFA. En lugar de ello, se
deberá emplear un IFA modelo de baja permeabilidad y un IFA modelo de alta
permeabilidad como patrones internos (incluidos en el fluido de perfusión o en
el fluido donante, junto con el IFA que se está estudiando). Estos dos patrones
internos se suman al marcador de volumen de fluido o de un compuesto de
permeabilidad cero como p. ej., PEG 4000, el cuál se incluye en ciertos tipos
de técnicas de perfusión (p. ej., técnicas de lazo cerrado). La selección de
patrones internos deberá basarse en la compatibilidad con el IFA en estudio
(esto significa que no deberán presentar ninguna interacción física, química o
de permeabilidad significativa). Cuando no sea factible cumplir este protocolo,
se deberá determinar la permeabilidad de los patrones internos del mismo modo
como se realiza la evaluación de permeabilidad del IFA en estudio, en un ensayo
paralelo, sobre los mismos sujetos, animales, tejidos o monocapas celulares.
Los valores de permeabilidad de los dos patrones internos no deberán diferir
significativamente en las distintas pruebas, incluyendo las que se realizan
para demostrar la aptitud del método. Al final de una prueba in situ o in
vitro, se recomienda determinar la cantidad de IFA en la membrana.
Para un método de prueba
determinado, en condiciones experimentales fijas, es posible que el empleo de
un patrón interno de alta permeabilidad o cercano al límite del tipo de
permeabilidad baja/alta (p. ej., metoprolol), facilite la clasificación del IFA
en estudio. Por ejemplo, se podrá determinar que el IFA en estudio es altamente
permeable cuando su valor de permeabilidad sea igual o mayor que el metoprolol
(IFA punto de corte).
3.1.1.2.3 Inestabilidad en
el sistema gastrointestinal
Algunos métodos empleados
para determinar la cantidad absorbida de un IFA no tienen en cuenta el alcance
de su degradación intestinal, antes de ingresar a la membrana del intestino.
Como ejemplo podemos citar: estudios en humanos (balance de masa) que se basan
en determinar la radioactividad total del IFA administrado, una vez que llega a
la orina; estudios de permeabilidad que se basan en la pérdida o la depuración
(clearence) del IFA desde los fluidos introducidos por perfusión en el sistema
gastrointestinal humano y/o animal ya sea in vivo o in situ.
Además de los procesos de
degradación, la pérdida del IFA desde el sistema gastrointestinal puede
producirse por la retención del mismo en la membrana intestinal.
Por lo expuesto, para
establecer con certeza la permeabilidad de un IFA, es necesario tener en cuenta
los hechos arriba mencionados.
Se deberá documentar la
estabilidad del IFA en el sistema gastrointestinal usando fluidos
gastrointestinales de modelos animales apropiados y/o fluidos simulados como
los Fluidos Gástrico e Intestinal USP a menos que sea posible disponer de
fluidos gástricos e intestinales obtenidos de sujetos humanos.
Se deberán incubar los
productos farmacéuticos en estos fluidos a 37°C durante un período que sea
representativo del contacto del IFA in vivo con estos fluidos; por ejemplo, 1
hora en fluido gástrico y 3 horas en fluido intestinal. Luego se deberán
determinar las concentraciones del IFA usando un método analítico validado que
indique la estabilidad. Una degradación significativa (> 5%) del IFA en este
estudio podría sugerir una posible inestabilidad.
3.1.2 Metodología para
determinar las características cinéticas de disolución de los productos
multifuente en estudio.
Para la bioexención de un
estudio de BE basado en el SCB, tanto el producto multifuente como el producto
comparador de referencia, deben exhibir características de disolución in vitro
muy rápidas o rápidas, dependiendo de la clase SCB a la que pertenezca el IFA.
Adicionalmente, podrá ser requerido que ambos productos presenten similares
perfiles de disolución.
3.1.2.1 Demostración de
Muy Rápida Cinética de Disolución.
Se debe demostrar que el
producto multifuente y el producto comparador de referencia liberan una
cantidad = 85% del IFA respecto del valor declarado a los 15 minutos, empleando
el Aparato II.FA.7ma a 75 rpm o el Aparato I.FA.7ma a 100 rpm, en un volumen de
900 mL o menos, en cada uno de los siguientes medios:
(1) Solución reguladora de
pH 1,2;
(2) Solución reguladora de
Acetato a pH 4,5 y
(3) Solución reguladora de
Fosfato a pH 6,8.
También son aceptables
soluciones reguladoras alternativas con el mismo pH y capacidad buffer, siempre
y cuando se demuestre que éstos no afectan algunas de las propiedades
fisicoquímicas del medio (p. ej., fuerza iónica) que puedan alterar la cinética
de disolución del IFA desde el producto.
3.1.2.2 Demostración de
Rápida Cinética de Disolución
Se debe demostrar que el
producto multifuente y el producto comparador de referencia liberan una
cantidad = 85% del IFA respecto del valor declarado a los 30 minutos, empleando
el Aparato II.FA.7ma a 75 rpm o el Aparato I.FA.7ma a 100 rpm, en un volumen de
900 mL o menos, en cada uno de los tres medios mencionados en el punto 3.1.2.1.
3.1.3 Productos a utilizar
en el estudio
En un estudio de
equivalencia basado en el SCB la diferencia en el contenido del IFA entre
producto multifuente y el producto comparador de referencia no debe ser mayor a
+/- 5 (Farmacopea Argentina 7ma edición, capítulo <740>)
3.1.3.1 Producto de
referencia
El producto de referencia
seleccionado deberá ajustarse a los requisitos estipulados en la Disposición
ANMAT N° 1918/13 o la/s que en el futuro la complemente o modifique. El lote
presentado deberá tener, al momento del estudio, por lo menos 6 meses hasta la
fecha de vencimiento.
3.1.3.2 Producto
multifuente
El laboratorio titular
deberá presentar tres lotes independientemente elaborados. Los mismos deben ser
iguales a aquellos a comercializarse, en cuanto a fórmula cuali-cuantitativa,
calidad de las materias primas, envase primario, método de elaboración, sitio
de elaboración, equipos empleados en la elaboración, y método analítico. El
tamaño de los lotes y la validación de los procesos productivos deben ajustarse
a lo establecido en la Disposición ANMAT N° 1263/12 (o la/s que en el futuro la
complemente o modifique), y estar elaborados bajo las Buenas Prácticas de
Fabricación y Control vigentes.
3.2 Bioexenciones basadas
en formulaciones proporcionalmente similares
3.2.1 Definición de
Formulaciones Proporcionalmente Similares
A los fines de solicitar
la bioexención, las formulaciones proporcionalmente similares deben ser
manufacturadas en el mismo sitio de elaboración y con los mismos procesos que
el producto que haya demostrado bioequivalencia/equivalencia in vitro basado en
el SCB.
Para las solicitudes de
bioexenciones por proporcionalidad, la composición de la/las dosis, deberán ser
proporcionalmente similares al producto que haya demostrado BE.
Dos formulaciones
proporcionalmente similares pueden ser definidas de dos formas complementarias:
a) Todos los ingredientes
de la formulación están exactamente en las mismas proporciones en las
diferentes dosis (p. ej., un comprimido de 50 mg de potencia y que posee una
cantidad de ingredientes inactivos que es exactamente la mitad de los
ingredientes inactivos que posee un comprimido de 100 mg, y el doble de los de
un comprimido de 25 mg).
b) En casos donde la
cantidad del IFA en el producto es relativamente baja (hasta 10 mg por unidad o
no más del 5% del peso total de esa forma farmacéutica). El peso total de la
forma farmacéutica se mantiene prácticamente igual para todas las dosis (dentro
de ± 10% del peso total), empleando los mismos ingredientes inactivos.
En el caso b) una
bioexención por proporcionalidad podrá ser considerada cuando:
- Las cantidades de los
diferentes excipientes son los mismos para las distintas dosis consideradas y
sólo la cantidad del IFA ha cambiado.
- La cantidad de relleno
se ha modificado, como consecuencia del cambio en la cantidad de IFA y las
cantidades de los otros excipientes son las mismas para las distintas dosis
consideradas.
3.2.2 Bioexención por
proporcionalidad según la forma farmacéutica
3.2.2.1 Comprimidos o
cápsulas de liberación inmediata
La bioexención de las
diferentes dosis de un producto multifuente puede ser considerada cuando:
(I) Las composiciones de
las distintas dosis son cuantitativas proporcionales.
(II) Se ha establecido la
bioequivalencia/equivalencia in vitro según corresponda para una de las dosis,
contra el producto de referencia. Generalmente la dosis estudiada es la mayor,
a menos que, por una razón de seguridad se haya establecido la bioequivalencia
con una dosis menor o que el IFA sea altamente soluble y posea farmacocinética
lineal.
(III) Los perfiles de
disolución de la dosis de referencia y la dosis a aprobar, son similares en los
tres medios especificados anteriormente y en el medio de control de calidad. En
caso de liberarse más de 85% a los 15 minutos, no es necesaria la comparación
de perfiles mediante el factor de similaridad (f2).
Si al comparar las
distintas dosis se ve afectada la condición sink, se deberá utilizar como
producto comparador al producto de referencia utilizado en los estudios de
bioequivalencia de igual dosis que el producto multifuente.
Cuando el producto
multifuente ha demostrado equivalencia con el producto de referencia basándose
en el SCB, la/s nueva/s dosis proporcionales deberán demostrar equivalencia con
el producto comparador de referencia de igual dosis.
3.2.2.2 Comprimidos o
cápsulas de liberación retardada
Para comprimidos de liberación
retardada, cuando el producto está en igual forma farmacéutica, las distintas
dosis poseen el mismo mecanismo de liberación, los mismos ingredientes y su
composición cuantitativa es proporcionalmente similar, una dosis menor a la
dosis con bioequivalencia in vivo aprobada puede ser exceptuada de presentar un
estudio de bioequivalencia si tiene un perfil de disolución similar (f2=50) en
las condiciones usuales para formas farmacéuticas de liberación retardada (2
horas en medio ácido, seguido de disolución en solución reguladora de pH 6.8).
Al evaluar la
proporcionalidad en la composición, se recomienda considerar la
proporcionalidad del recubrimiento gastroresistente con respecto a la
superficie del comprimido (no del peso del núcleo) para tener el mismo grado de
gastrorresistencia (mg/cm2).
En el caso de cápsulas,
cuando la diferencia de las dosis es la consecuencia de una disminución en la
cantidad de gránulos gastroresistentes que contienen el IFA, la similaridad
(f2=50) entre el perfil de disolución de la nueva dosis (menor) y la dosis con
bioequivalencia aprobada, en las condiciones usuales de disolución para formas
farmacéuticas de liberación retardada, será suficiente para sustentar una
bioexención.
3.2.2.3 Cápsulas de
liberación prolongada
Para cápsulas de
liberación prolongada, cuando la diferencia de dosis es la consecuencia
únicamente de una disminución en la cantidad de gránulos que contienen el IFA,
la similaridad (f2=50) entre el perfil de disolución de la nueva dosis (menor)
y la dosis con bioequivalencia aprobada, realizada en las condiciones de
disolución aprobadas para el producto terminado, será suficiente para sustentar
una bioexención.
3.2.2.4 Comprimidos de
liberación prolongada
Cuando la dosis menor
tiene los mismos ingredientes, su composición cuantitativa es proporcionalmente
similar, y tiene el mismo mecanismo de liberación, podrá justificarse una
bioexención si demuestra similaridad con la dosis con bioequivalencia aprobada,
en los perfiles de disolución (f2=50) de por lo menos tres medios de disolución
diferentes, de pH entre 1.2 y 7.5 y el medio Cuando la dosis menor tiene los
mismos ingredientes, su composición de control de calidad.
3.2.2.5 Farmacocinética
lineal
En todos los casos, el
solicitante de la Bioexención deberá demostrar o referenciar, además de la
proporcionalidad entre las formulaciones, que el IFA posee farmacocinética
lineal en el rango de concentraciones terapéuticas. En el contexto de esta
guía, se considera farmacocinética lineal cuando un incremento de la dosis
resulta en un incremento proporcional del área bajo la curva (AUC). En caso de
IFAS con farmacocinética no lineal la sensibilidad del método de disolución
puede no ser suficiente para detectar diferencias entre las distintas dosis.
3.2.3 Productos a utilizar
en el estudio
Dependiendo del rango
terapéutico del IFA, se deberá usar al menos un lote para IFAs de amplio rango
terapéutico y al menos dos lotes para IFAS de estrecho margen terapéutico.
3.2.3.1 Producto de
referencia
Es el producto cuya
titularidad pertenece al mismo laboratorio; generalmente el de dosis más alta
que el que se presenta para aprobar, y para el cual la ANMAT ha aceptado la
demostración de bioequivalencia con el producto de referencia establecido. En
caso que la equivalencia (generalmente el de dosis más alta) haya sido
demostrada mediante ensayos in vitro se empleará el producto de referencia, de
igual dosis que el producto multifuente, que se ajuste a los requisitos
estipulados en la Disposición ANMAT N° 1918/13 o la/s que en el futuro la
complemente o modifique.
Deberá tener al momento
del estudio, por lo menos 6 meses hasta la fecha de vencimiento. Es
recomendable la presentación de resultados de valoración y cinética de
disolución de más de un único lote del producto de referencia.
3.2.3.2 Producto
multifuente
En la solicitud de
bioexención, el laboratorio titular deberá presentar lote/s elaborado/s bajo
las Buenas Prácticas de Fabricación y Control (Disposición ANMAT N° 2819/2004 o
la/s que en un futuro la complemente o modifique).
El tamaño de los lotes y
la validación de los procesos productivos deben ajustarse a lo establecido en
la Disposición ANMAT N° 1263/12 (o la/s que en un futuro la complemente o
modifique).
3.3 Bioexenciones para
cambios posteriores a demostración de bioequivalencia
Ciertos cambios menores en
la elaboración, de formas farmacéuticas sólidas orales de liberación inmediata,
pueden ser autorizados sin riesgo de bioinequivalencia con el biolote, según se
establece en la Disposición ANMAT N° 556/09.
3.3.1 Productos a analizar
en el estudio
3.3.1.1 Producto de
referencia
El lote del producto de
referencia presentado deberá tener, al momento del estudio, por lo menos 6
meses hasta la fecha de vencimiento. Es recomendable la presentación de
resultados de valoración y cinética de disolución de más de un único lote del
producto de referencia. (17)
3.3.1.2 Producto
multifuente en estudio
El producto elaborado en
las condiciones y/o composición propuestas. El número de lotes dependerá del
nivel de cambio aprobado.
4. CONSIDERACIONES
ADICIONALES PARA SOLICITAR UNA BIOEXENCIÓN
Cuando se solicita una
bioexención basándose en el SCB para los estudios de BD/BE in vivo de FFSO-LI,
los solicitantes deberán tener en consideración las siguientes situaciones
particulares que pueden afectar su solicitud o la documentación que deberán
adjuntar a ésta:
4.1 Excipientes
Algunos excipientes pueden
afectar la velocidad y la cantidad de IFA absorbido. Para respaldar una
solicitud de bioexención la cantidad de excipientes en el producto farmacéutico
de liberación inmediata deberá corresponder a la función prevista (p. ej.,
lubricantes).
Se deberá establecer la
adecuabilidad de los excipientes aportando documentación correspondiente a
cualquiera de las siguientes alternativas:
1- El/los excipiente/s
están presentes en cantidades similares en el Producto Comparador de
Referencia.
2- El /los excipientes
están presentes en cantidades similares en productos que contienen el mismo
principio activo y poseen autorización de comercialización en países miembros
de ICH.
3- El /los excipientes
están presentes en el producto en cantidades normalmente empleadas para esa
forma farmacéutica. Consultar en el sitio:
www.accessdata.fda.gov/scripts/cder/iig/index.cfm de la FDA para búsqueda de
ingredientes inactivos utilizados en productos aprobados.
Cuando se incluyen excipientes
nuevos o cantidades atípicamente grandes de excipientes de uso común en una
forma farmacéutica sólida de liberación inmediata (p. ej., agentes
tensoactivos), la ANMAT podrá pedir información adicional que documente la
ausencia de un posible impacto sobre la BD del IFA. En determinadas ocasiones,
tal información puede proporcionarse con un estudio de BD relativa usando una
solución acuosa simple como producto de referencia.
Ciertos excipientes en
cantidades inusuales, como surfactantes (p. ej., polisorbato 80) y edulcorantes
(p. ej., manitol o sorbitol), podrían ser cuestionables al provocar un posible
impacto sobre la BD de IFA. Para las formulaciones que contengan algún/os de
estos excipiente/s deben ser cualitativamente los mismos y cuantitativamente
similares a aquellos del producto de referencia. (18)
Para formulaciones que
solicitan bioexención por proporcionalidad de dosis, la información sobre
excipientes, se complementa en el punto 3.2.1 de la presente guía.
Para formulaciones basadas
en el SCB que contengan IFAS de clase I, podrá permitise cierta flexibilidad en
los excipientes empleados con excepción de los excipientes críticos mencionados
anteriormente. (18-20)
Para formulaciones que
contengan IFAS de clase III, los excipientes debe/n ser cualitativamente los
mismos y cuantitativamente similares a aquellos del producto de referencia.
(18-20)
La cantidad relativa de
excipientes en dos formas sólidas orales se considera cuantitativamente similar
si las diferencia en las cantidades de excipientes se encuentran dentro de los
límites descriptos en la tabla 3.
Tabla 3: Diferencia
aceptable, expresada como porcentaje (p/p) de la formulación total del
producto, para que los excipientes de dos formas farmacéuticas sean
considerados cuantitativamente similares.
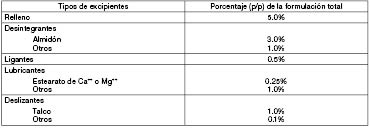
4.2 Profármacos
La permeabilidad de los
profármacos dependerá del mecanismo y del sitio (anatómico) de su conversión en
el fármaco propiamente dicho. Cuando se demuestra que la conversión de
profármaco en fármaco ocurre predominantemente después del pasaje de la membrana
intestinal, se deberá medir la permeabilidad del profármaco. Cuando esta
conversión ocurre antes del pasaje de la membrana intestinal, se deberá
determinar la permeabilidad del fármaco. Los datos sobre la disolución, así
como también de solubilidad en función del pH, para el profármaco pueden ser
relevantes. Para optar a una bioexención, los patrocinadores podrían considerar
conveniente consultar con el personal de la agencia regulatoria, antes de
solicitar el empleo del método del SCB para productos de liberación inmediata
que contienen profármacos.
4.3 Situaciones en las que
no aplican las bioexenciones
Las bioexenciones en base
al SCB no son aplicables para los siguientes productos farmacéuticos:
4.3.1 Productos de
estrecho margen terapéutico
Esta guía define a los
productos de estrecho margen terapéutico como aquellos que contienen ciertos
IFAS que están sujetos a control de la concentración de IFA (monitoreo
terapéutico) o a monitoreo farmacodinámico, y/o donde la información científica
farmacológica disponible del IFA indica que presenta estrecho margen
terapéutico. Los ejemplos incluyen: digoxina, litio, fenitoína, teofilina y
warfarina. Ya que no siempre todos los IFAS que están sujetos a monitoreo
terapéutico o a monitoreo farmacodinámico son IFAS de productos de estrecho
margen terapéutico, en algunas situaciones los patrocinadores deberán
contactarse con la agencia regulatoria, para determinar si se debe considerar
que un determinado IFA corresponde efectivamente a uno de estrecho margen
terapéutico.
4.3.2. Productos diseñados
para ser absorbidos en la cavidad oral
Para formas farmacéuticas
diseñadas para absorción en la cavidad oral (p. ej., comprimidos sublinguales o
bucales), no es apropiada una solicitud de bioexención de estudios de BD/BE in
vivo en base al SCB.
5- CONDICIONES
EXPERIMENTALES DE LOS ESTUDIOS CINÉTICOS COMPARATIVOS DE DISOLUCIÓN PARA
DEMOSTRAR EQUIVALENCIA.
5.1 Equipos y
Procedimientos
Los ensayos de disolución
deben realizarse en el aparato de paletas, (Aparato II.FA.7ma), a 75 rpm o en
el aparato de canastillo (Aparato I.FA.7ma) a 100 rpm, en un volumen de 900 mL
o menos, en los tres medios de disolución mencionados en 3.1.2.1. Los medios de
disolución deben desgasificarse inmediatamente antes de comenzar el ensayo,
filtrando cada medio al vacío y calentado previamente a 41°C, utilizando un
filtro de membrana de 0,45 µm con agitación vigorosa. Puede emplearse otra
metodología de desgasificado, adecuadamente validada.
Los equipos y
procedimientos generales deberán estar de acuerdo con lo establecido en la
Farmacopea Argentina o la USP edición vigente, así como con las siguientes
recomendaciones:
a) No resulta admisible el
comienzo escalonado del ensayo de disolución.
b) En el aparato de
paletas, las unidades deben ser introducidas en cada vaso con el equipo
detenido e inmediatamente después comenzar la agitación. En el aparato de
canastillo, las muestras deben colocarse en los cestillos perfectamente secos,
introducir los cestillos en los vasos, e inmediatamente después iniciar la
agitación.
c) La extracción de las
muestras puede realizarse en modo manual o automático. Si es manual, deberá
usarse una jeringa con cánula de acero provisto de un filtro de tamaño de poro
de entre 0.45 y 0.70 µm que puede estar ubicado en el extremo de la cánula o en
su unión con la jeringa. Debe tenerse en cuenta la tolerancia máxima admitida
en el tiempo de muestreo entre el primer vaso y el último. El rango admitido
del tiempo de muestreo es ± 2% resultando, p. ej., para el muestreo a los 10
minutos (600 segundos), una diferencia entre el primer y último vaso de 12
segundos. Si no es posible la toma de muestra dentro de este margen de
tolerancia, deberá usarse muestreo automático. En este caso deberá validarse la
ausencia de interferencia del muestreo automático en los resultados.
d) No son admisibles para
el filtrado de las muestras el filtrado por papel ni centrifugación.
Para calificar al equipo
de disolución se recomienda realizar:
-Calibración mecánica: Con
una antelación no mayor a seis meses de realizar los estudios comparativos de
disolución, se recomienda verificar los siguientes parámetros críticos del
equipo de disolución: horizontalidad, centrado y vaivén de los ejes, vaivén del
canastillo, fluctuaciones de temperatura, velocidad de rotación de los ejes y
altura del canastillo. Los límites aceptables son los establecidos en la USP
vigente.
-Calibración química: Se
recomienda emplear los comprimidos USP Prednisone tablets RS, ajustándose a las
especificaciones establecidas para el lote vigente.
5.2 Validación del método
analítico empleado en el estudio de disolución
Para que los resultados
experimentales sean confiables, los métodos utilizados deberán ser
apropiadamente validados. Se entiende por validación, al proceso mediante el
cual se demuestra la aplicabilidad de un método analítico y consiste en el
establecimiento de una evidencia documentada que demuestre con alto grado de
probabilidad que el método es confiable para producir el resultado previsto
dentro de los intervalos definidos. (19)
Los parámetros
recomendados para la validación del método analítico utilizado son: linealidad,
exactitud, precisión y robustez. Además se recomienda documentar la especificidad
del método, la influencia del muestreo automático y del sistema de filtración
de las muestras extraídas desde los vasos de disolución, la estabilidad del
analito en las soluciones, la precisión intermedia y los límites de
cuantificación y detección del analito. (19)
5.2.1 Especificidad
Es la capacidad de un
método de cuantificar el analito en presencia de otros componentes que puedan
estar presentes (impurezas, productos de degradación, aditivos). Para ello se
recomienda:
• Pesar por triplicado
muestras placebo (colorantes, excipientes, cápsulas vacías etc.), a
concentración equivalente a la que contendría la dosis mayor y menor.
• Transferir la mezcla a
vasos conteniendo medio de disolución a 37°C y agitar durante 1 hora a 150 rpm.
• Analizar las muestras.
• Calcular el porcentaje
de interferencia a cada una de las concentraciones (n = 3), comparando con una
solución estándar al 100% de la dosis.
La fórmula para calcular
la interferencia en el método analítico es:
Donde:
I = Interferencia (en %)
C = Concentración de la
solución estándar (mg/mL)
Ap y As = Absorbancia o
Áreas del placebo y del estándar
V = Volumen de medio en el
vaso (mL)
L = Dosis del producto,
según rótulo (mg)
Especificación: La
interferencia media no debe exceder el 2%
Si no se conoce la
composición del placebo, se recomienda realizar la disolución de la formulación
y analizarla. Luego, comparar la respuesta del compuesto a analizar contra una
solución patrón o de referencia. Si el método es cromatográfico, se recomienda
evaluar la interferencia de los picos en el tiempo de retención del analito.
5.2.2 Linealidad y Rango
Rango es el intervalo
entre la concentración superior e inferior de concentración del analito en la
muestra que ha demostrado que el método presenta un nivel adecuado de
precisión, exactitud y linealidad. El rango de concentraciones se calcula de
los estudios de linealidad.
• Si el método es
espectrofotometría UV: determinar la longitud de onda máxima de absorción.
• Preparar una solución al
100% y determinar su respuesta (Absorbancia).
• Verificar si hay
necesidad de diluir.
• Verificar que el medio
no absorba a la misma longitud de onda.
• Si el método es por
HPLC, se debe verificar que el medio no interfiera en el tiempo de retención
del analito.
• Preparar una curva
equivalente entre 10-120% de IFA disuelto.
Linealidad es la capacidad
de un método analítico de asegurar que los resultados obtenidos son
directamente proporcionales a la concentración del analito en la muestra. Para
ello:
• Se prepara la curva de
calibración.
• Se calculan el
coeficiente de correlación, el intercepto, la pendiente y la suma de cuadrados
residuales.
• El coeficiente de
correlación no deberá ser menor a 0,98, el intercepto en “y” no debe ser
diferente de 0, empleando un intervalo de confianza de 95%.
• El coeficiente de
variación del factor de respuesta no debe ser mayor del 2%.
5.2.3 Exactitud
Expresa la cercanía entre
el valor verdadero y el valor que es aceptado (sea como un valor convencional
verdadero o un valor de referencia aceptado). La definición práctica indica que
corresponde a la relación estrecha que existe entre la media aritmética
obtenida de varios resultados analíticos en el procedimiento en estudio y el
valor real o de referencia, lo cual está influenciado por errores sistemáticos
como, por ejemplo, equipos de medición inadecuados, calidad incorrecta de los
reactivos y método analítico inapropiado. Para ello se recomienda:
• Pesar el IFA (por
triplicado) para obtener 3 concentraciones conocidas (incluyendo
concentraciones mínima y máxima del intervalo).
• Añadir el IFA a vasos de
disolución conteniendo el placebo.
• Analizar las muestras.
• Criterio: Una
recuperación del 95-105% es aceptable. La desviación estándar relativa deberá
ser =2%.
5.2.4 Precisión del método
Expresa la coincidencia
(grado de dispersión) entre una serie de múltiples muestreos de una misma muestra
homogénea, bajo condiciones establecidas. Corresponde al grado de concordancia
de los datos obtenidos para una muestra procesada varias veces, lo que está
limitado por errores aleatorios como los errores instrumentales e individuales,
entre otros. Para ello se recomienda:
• Analizar réplicas de una
solución estándar (n = 6).
• Calcular la desviación
estándar relativa.
• Criterio: La desviación
estándar relativa no deberá ser mayor al 2%.
5.2.5 Precisión Intermedia
Expresa la variación en
los resultados observados cuando uno o más factores, tales como tiempo,
equipamiento, operador, varían dentro de un mismo laboratorio. Dos analistas
diferentes realizan dos estudios cinéticos de disolución (n=12) y se comparan
los resultados promedio y los coeficientes de variación (%). La diferencia en
los valores promedio, no debe exceder un 10% en aquellos tiempos en los que se
ha disuelto = 85% y un 5% en los que se ha disuelto más del 85% del IFA en
estudio.
5.2.6 Robustez
Se debe determinar durante
la fase de desarrollo del método. Corresponde a una medida de la capacidad de
un procedimiento analítico de entregar resultados analíticos con exactitud y
precisión aceptables, frente a cambios pequeños, pero deliberados, en los
parámetros del método (por ej., en un método espectrofotométrico, frente a
pequeños cambios en la longitud de onda). Para ello, se recomienda realizar
cambios pequeños, pero deliberados a las condiciones de disolución (n = 3).
Dependiendo del método, los cambios pueden ser los siguientes:
• Soluciones reguladoras,
modificar pH en 0,5 unidades arriba o abajo.
• HPLC: cambios de
columna, flujo de la fase móvil, pH de la fase móvil, etc.
• Espectrofotometría UV:
cambios en la longitud de onda ± 2 nm.
5.2.7 Estabilidad
Se debe asegurar la
estabilidad de la solución estándar y de las muestras hasta el momento de su
análisis. Para determinar la estabilidad de la solución estándar se recomienda
almacenarla en condiciones que aseguren su estabilidad (por ej. en refrigeración),
analizarla en el período de tiempo especificado y comparar el resultado con el
obtenido con una solución estándar recientemente preparada. El rango de
recuperación deberá estar entre 98-102% del valor promedio. Para el caso de las
muestras se debe proceder de la misma forma. Vale decir, almacenar la solución
en condiciones que aseguren su estabilidad, analizarla en el tiempo establecido
y comparar el resultado con el obtenido inmediatamente de extraída la muestra
desde el vaso de disolución. El porcentaje de recuperación deberá encontrarse
entre 98-102% del valor promedio.
5.2.8 Interferencia del
Filtro
Se analiza una solución de
concentración conocida, luego se filtra y se mide nuevamente. Se comparan ambos
resultados, siendo aceptable un rango de recuperación entre 98-102%.
5.2.9 Validación del
muestreo automatizado
Se construyen dos perfiles
de disolución con 6 unidades posológicas, muestreando simultáneamente de manera
manual y automática, y se comparan los resultados obtenidos usando el criterio
indicado en 5.2.5 Precisión Intermedia.
6. DOCUMENTACIÓN NECESARIA
PARA RESPALDAR UNA SOLICITUD DE BIOEXENCIÓN
6.1 Solicitud de
aprobación de Protocolo de Bioexención y Diseño de los estudios in vitro.
Presentación de resultados
Previo a la realización de
los estudios in vitro (solubilidad, permeabilidad y cinética de disolución),
los interesados deberán presentar al INAME la solicitud de bioexención
correspondiente.
Se deberán completar los
siguientes anexos según el caso (ver al final de la presente guía):
-Anexo I: “SOLICITUD DE
BIOEXENCIÓN según DISPOSICIÓN ANMAT N° 758/09. Bioexención basada en la
Clasificación Biofarmacéutica”.
-Anexo II: “SOLICITUD DE
BIOEXENCIÓN según DISPOSICIÓN ANMAT N° 758/09. Bioexención basada en
formulaciones proporcionalmente similares”.
-Anexo III: “SOLICITUD DE
BIOEXENCIÓN según DISPOSICIÓN ANMAT N° 556/09. Cambios posteriores a la
aprobación del registro de un producto farmacéutico que ha demostrado
Bioequivalencia”.
En todos los casos, la
ANMAT procederá a autorizar la realización de dichos estudios, si considera
adecuada la información aportada. En caso de que la solicitud no corresponda,
el trámite será denegado.
Las áreas involucradas en
la evaluación de la solicitud de bioexención serán las responsables de otorgar
la conformidad o de señalar los cambios que se estimen pertinentes en los
protocolos de estudio. La ANMAT, autorizará al laboratorio Solicitante a llevar
a cabo la demostración de equivalencia por metodología in vitro. En
consecuencia, éstos sólo podrán realizarse una vez extendida la aprobación
oficial de los protocolos por parte de la ANMAT.
Para presentar los
resultados obtenidos se deberán completar los anexos correspondientes según el
caso (ver al final de la presente guía):
-Anexo IV: “PRESENTACIÓN
DE RESULTADOS PARA SOLICITAR UNA BIOEXENCIÓN SEGÚN DISPOSICIÓN ANMAT N° 758/09.
Bioexención basada en la Clasificación Biofarmacéutica”.
-Anexo V: “PRESENTACIÓN DE
RESULTADOS DE PROPORCIONALIDAD DE DOSIS PARA SOLICITAR UNA BIOEXENCIÓN SEGUN
DISPOSICIÓN ANMAT N° 758/09”.
-Anexo VI: “DOCUMENTACIÓN
REQUERIDA PARA LA PRESENTACIÓN DE RESULTADOS DE CAMBIOS POSTERIORES A LA
APROBACIÓN DE UN REGISTRO PARA SOLICITAR UNA BIOEXENCIÓN SEGUN DISPOSICIÓN
ANMAT N° 556/09”.
Las áreas involucradas en
la evaluación de los resultados serán las responsables de otorgar la
conformidad o de requerir al laboratorio documentación, pruebas adicionales, o
demostración de equivalencia in vivo, según corresponda.
En caso de resultar
aceptable la información aportada en el trámite, la ANMAT aceptará la
bioexención, mediante una Disposición aprobatoria.
6.2 Información sobre el
laboratorio y el producto farmacéutico multifuente para el cual se solicita la
bioexención
6.2.1 Datos del
laboratorio solicitante
El interesado deberá
completar los requisitos correspondientes del formulario de la solicitud de
bioexención.
6.2.2 Requisitos sobre el
producto multifuente en estudio
El interesado deberá
completar los requisitos correspondientes del formulario de la solicitud de
bioexención.
En los casos de formulaciones
no registradas, la autorización para la realización de los estudios de
bioexención no se efectivizará hasta que, las áreas técnicas correspondientes
del ANMAT, hayan evaluado y aceptado la solicitud de registro. En los casos de
formulaciones previamente registradas, deberá incluirse en la solicitud de
bioexención el número de certificado correspondiente.
6.2.3 Establecimiento/s
propuesto/s para realizar los estudios del Producto/s, y en caso de
corresponder, de los IFA
Para cada uno de los
ensayos de solubilidad, permeabilidad y cinética de disolución, ya sean
realizados por el solicitante o tercerizados, se deberán completar los ítems
mencionados en el formulario correspondiente.
6.2.4 Caracterización de
elaboración y Buenas Prácticas de Fabricación y Control.
6.2.4.1 Caracterización de
elaboración
Para el IFA en estudio se
deberá presentar documentación que avale:
a) El cumplimiento de las
especificaciones de calidad y consistencia en resultados obtenidos con los
lotes propuestos para los estudios de bioequivalencia o equivalencia in vitro
del producto multifuente.
b) Se deberá declarar la
función que cumple cada excipiente en la formulación, dado que existe evidencia
científica de que algunos excipientes pueden modificar la motilidad y/o
permeabilidad del tracto gastrointestinal. De la misma forma, se deberá
presentar un cuadro comparativo con la composición cuali/cuantitativa de los
productos de referencia y multifuente, a los fines de establecer la
adecuabilidad de los excipientes según ítem V Excipientes de la Disposición
ANMAT N° 758/09.
6.2.4.2 Caracterización de
Buenas Prácticas de Fabricación y Control
Para el IFA en estudio se
deberá presentar documentación que avale que el proceso de elaboración ha sido
validado y que la fabricación del producto se ha realizado siguiendo las Buenas
Prácticas Fabricación y Control vigentes (Disposición ANMAT N° 2819/2004 o la/s
que en el futuro la complemente o modifique).
6.3 Datos que respaldan la
alta solubilidad del IFA
Se deberán presentar
resultados respaldando la alta solubilidad del IFA detallando:
• Descripción del método
analítico, la validación del mismo y composición de soluciones reguladoras.
• Información acerca del
IFA, detallando estructura química, el peso molecular, naturaleza química
(ácido, base, anfótero o neutro), constante de disociación y valores de los
coeficientes de partición y distribución del mismo.
• Representación gráfica
de los resultados de solubilidad en un gráfico solubilidad vs pH.
• Resultados individuales
de solubilidad, indicando el pH de la solución empleada, expresados en unidades
de concentración (mg/mL) y el volumen del medio requerido para disolver la
mayor concentración posológica. Resultados promedios. Desviación estándar.
6.4 Datos que respaldan la
alta permeabilidad del IFA en caso de que no esté en el listado de la OMS
Se deberán presentar
resultados propios y antecedentes científicos válidos que avalen la
permeabilidad del IFA utilizado en la elaboración del producto farmacéutico.
En la solicitud, se deberá
incluir la siguiente información:
- En caso de utilizar
métodos de permeabilidad en humanos, se debe aportar información acerca del
diseño del estudio y de los métodos farmacocinéticos, analíticos y
bioestadísticos utilizados, junto con los datos farmacocinéticos “crudos” del
estudio. De acuerdo con los requisitos enumerados en: Disposiciones ANMAT N°
6677/10, 3185/99, 3598/02, 5040/06 y la/s que en el futuro la/s modifiquen o
complementen.
- En caso de utilizar
métodos de permeabilidad alternativos, se debe presentar descripción del método
de estudio de permeabilidad a emplear, criterios de selección de animales o
línea celular, concentraciones del IFA a emplear en fluido donante e informe de
validación del método analítico.
Se debe entregar en
detalle el listado de IFAS modelo utilizados para la Validación del método de
estudio de la permeabilidad, los valores de permeabilidad (promedio, desviación
estándar y CV%), y un gráfico de fracción absorbida en humanos en función de la
permeabilidad, identificando el límite de permeabilidad alta/baja y el patrón
interno seleccionado para este fin. El número de IFAS modelo deberá ser quince
(15). Esta lista deberá incluir IFAS de permeabilidad alta, moderada y baja,
además de un IFA punto de corte (p. ej., metoprolol) incluyendo en todos los
casos su respectiva clasificación biofarmacéutica.
6.5 Datos que respaldan la
cinética de disolución
En el trámite de solicitud
de bioexención de un producto, deberán presentar información sobre las
características del equipo de disolución utilizado (marca, equipo, calibración,
etc).
Por otro lado, basado en
el SCB del IFA, se deberá presentar información que respalde las
características de rápida o muy rápida disolución (punto 3.1.2 de este
documento).
6.6 Evaluación de la
solicitud de bioexención
En caso de resultar
aceptable la información aportada en el trámite y la/las metodología/s
propuesta/s para los ensayos correspondientes, la ANMAT aceptará la solicitud
de bioexención, que autorizará al laboratorio solicitante a llevar a cabo la
demostración de equivalencia por metodología in vitro.
Si la solicitud de
bioexención es objetada, la ANMAT requerirá al laboratorio documentación,
pruebas adicionales o demostración de equivalencia in vivo, según corresponda.
6.7 Verificación técnica
En caso de tratarse de un
producto aún no comercializado, corresponderá realizar la verificación técnica
de acuerdo a la Disposición ANMAT N° 5743/09.
El laboratorio deberá
comunicar fehacientemente con 30 días de anticipación el inicio de los estudios
de cinética comparativa de disolución. El personal técnico de la ANMAT podrá
presenciar o de ser necesario, repetir o hacer repetir parcial o totalmente el
estudio comparativo. En caso de realizar los ensayos de solubilidad y
permeabilidad, se deberá comunicar con el mismo lapso de tiempo, el período en
el cual se realizarán dichos ensayos.
6.8 Retención de
Contramuestras
Se deberán mantener tres
contramuestras de los lotes de los productos de referencia y multifuente
utilizados en los ensayos, durante 2 años y 6 meses desde el momento de la
aprobación del estudio, respetando las condiciones de almacenamiento indicadas
en el rotulado. Cada contramuestra deberá contener al menos 60 unidades de cada
producto.
6.9 Presentación de
resultados
Los resultados de los
estudios de solubilidad, permeabilidad y del estudio de cinética de disolución
comparativa deberán presentarse de acuerdo a los formularios correspondientes,
independientemente de si el personal técnico de la ANMAT hubiera o no
presenciado los estudios.
Los resultados obtenidos
en los estudios comparativos deberán informarse de acuerdo a la solicitud
aprobada, completando en cada caso la tabla que corresponda en el Anexo VII:
“GUÍA DE BIOEXENCIONES. RESULTADOS DE LOS ESTUDIOS DE DISOLUCIÓN”. Se indicará
el porcentaje de IFA disuelto en cada intervalo de prueba, para cada unidad
posológica individual, consignándose el porcentaje promedio de IFA disuelto, el
rango (mayor y menor) de disolución y el coeficiente de variación (desviación
estándar relativa).
En la comparación de los
productos multifuente (M) y referencia (R), se deberá confrontar los perfiles
cinéticos de disolución usando el factor de similitud (f2); cuando esta
metodología no fuese adecuada pueden aplicarse metodologías estadísticas
alternativas siempre que las mismas sean válidas y justificadas
satisfactoriamente. El factor de similitud corresponde a la transformación
logarítmica de la raíz cuadrada recíproca de la suma del error cuadrado y
proporciona una estimación de la similitud en las cinéticas de disolución entre
ambos productos.
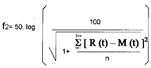
Los dos perfiles se
consideran similares cuando el valor de f2 es = 50.
Para permitir el uso de
datos promedio, el coeficiente de variación no deberá ser más del 20% en los
puntos temporales más tempranos del perfil de disolución (p.ej., 5 o 10
minutos), y no más del 10% en los demás puntos temporales del perfil.
Para el cálculo de f2 se
requiere un mínimo de tres puntos temporales (excluir el cero y tomar un solo
punto temporal en el que el valor de disolución para el producto de referencia
sea igual o mayor al 85%). Un valor de f2 de 50 o mayor (50-100) refleja la
equivalencia de las dos curvas.
Cuando el IFA contenido
tanto en el producto de referencia como en el producto multifuente en estudio,
se disuelve en un porcentaje de 85% o más de la cantidad declarada del IFA a
los 15 minutos o menos, utilizando los medios de disolución que correspondan,
la disolución de los productos es considerada equivalente y no es necesaria la
comparación de perfiles.
6.10 Aceptación o rechazo
de la Equivalencia
6.10.1 Bioexención basada
en el SCB del IFA
La ANMAT se expedirá sobre
la aceptación o rechazo de la equivalencia del producto presentado respecto al
producto de referencia.
6.10.2 Bioexención por
formulaciones proporcionalmente similares
La ANMAT se expedirá sobre
la aceptación o rechazo de la equivalencia del producto presentado respecto al
de referencia (diferente dosis del mismo producto con equivalencia aceptada).
6.10.3 Bioexención por
cambios post aprobatorios según Disposición ANMAT N° 556/09
La ANMAT se expedirá sobre
la aceptación o rechazo de la equivalencia del producto con los cambios
propuestos respecto al producto con equivalencia demostrada.
7. CÓMO TRAMITAR UNA
BIOEXENCIÓN
7.1 Protocolo del estudio
Se deberá completar el
Formulario de Solicitud de Bioexención correspondiente (Anexos I-III). La
información solicitada, se presentará en Mesa de Entradas del INAME.
Evaluada la información
por las áreas pertinentes, la aceptación del trámite de solicitud se hará
mediante nota aprobatoria de la Dirección Nacional.
7.2 Resultados del estudio
Con el protocolo de
solicitud aprobado, una vez terminado el estudio, se deberá completar el
Formulario de Presentación de Resultados para una Bioexención (Anexos IV-VII)
acompañado con la documentación correspondiente. Esta información deberá ser
presentada en Mesa de Entradas INAME para ser incorporada al expediente de
Bioexención.
7.3 Certificación de un
producto como Equivalente
Frente a la demostración
de equivalencia entre el producto multifuente y el producto comparador de
referencia, la ANMAT procederá a dictar la disposición correspondiente
declarando al producto multifuente PRODUCTO EQUIVALENTE al producto de
referencia.
8. DISPOSICIONES ANMAT
RELACIONADAS CON LA PRESENTE GUÍA
-5904/96
Definiciones y
lineamientos generales acerca del modo en que deberá incluirse la información
que deben contener los prospectos de especialidades medicinales cuya condición
de expendio sea la de venta bajo receta en sus tres categorías.
-3185/99
Apruébanse las
recomendaciones técnicas contenidas en el documento “cronograma para exigencias
de estudios de equivalencia entre medicamentos de riesgo sanitario
significativo”.
-3311/01
Establécense las
condiciones en las cuales deberán realizarse los estudios, de
bioequivalencia/biodisponibilidad de las especialidades medicinales que
contengan como principio activo individual uno de los antirretrovirales
utilizados para el tratamiento de la infección con el virus de la
inmunodeficiencia humana, tanto para las actualmente en comercialización, como
las registradas y no comercializadas.
-2807/02
Incorporación de a la
droga Isotretinoína al cronograma de bioequivalencia. Selección de productos
comparadores para estudios de bioequivalencia, para las drogas Carbamazepina,
Oxcarbazepina, Valproato, Ciclosporina, Teofilina, Verapamilo, Digoxina e
Isotretinoína.
-2819/04
Apruébense los
lineamientos generales de Buenas Prácticas de Fabricación para Elaboradores,
Importadores/Exportadores de Medicamentos.
-5040/06
Apruébase el Régimen de
Buenas Prácticas para la Realización de Estudios de
Biodisponibilidad/Bioequivalencia.
-1746/07
Sustitúyese el Anexo I de
la Disposición N° 5040/2006, referido al Régimen de Buenas Prácticas para la
Realización de Estudios de Biodisponibilidad/Bioequivalencia.
-2446/07
Incorpóranse determinados
principios activos (Serolimus, Everolimus, Tacrolimus y Micofenolato) a la
exigencia de realización de estudios de Bioequivalencia/Biodisponibilidad,
establecidos por la Disposición N° 3185/99.
-556/09
Apruébase la Guía para
aplicar en los Cambios de Escala y Cambios Posteriores al Registro de
Medicamentos Sujetos a Demostración de Bioequivalencia.
-758/09
Criterios de Bioexención
de Estudios de Bioequivalencia para Medicamentos sólidos orales de liberación
inmediata.
-5743/09
La evaluación de la
información relacionada con los métodos de control, elaboración, ensayos
farmacotécnicos, estudios de estabilidad, capacidad operativa para elaborar y/o
de control incluida en las solicitudes de inscripción en el registro de
especialidades medicinales, se realizara mediante la verificación técnica de
dicha información, debiendo cumplimentarse los requisitos establecidos en la
Disposición ANMAT N° 2819/2004 o la que en el futuro la reemplace y las
especificaciones de calidad establecidas en la farmacopea argentina, farmacopea
brasileña y/u otra farmacopea internacionalmente reconocida.
-6677/10
Apruébase el Régimen de
Buena Práctica Clínica para Estudios de Farmacología Clínica.
-3113/10
Incorpóranse a la exigencia
de realización de estudios de Bioequivalencia / Biodisponibilidad, establecidos
por la Disposición (ANMAT) N° 3185/99, a los ingredientes farmacéuticos activos
Lamotrigina y Topiramato
-1263/12
Establécese que, a los
fines de la realización del estudio de bioequivalencia o equivalencia “in
vitro”, según corresponda, el laboratorio patrocinante deberá proponer alguna
de las tres opciones siguientes: a) tres lotes vigentes, de escala industrial,
idénticos al producto a comercializarse, b) tres lotes pilotos de tamaño no
menor a las 100.000 unidades o c) tres lotes pilotos sin establecer un tamaño
mínimo de unidades; las opciones b) o c) solo serán aceptables siempre que los
lotes se elaboren en equipos de escala industrial que se encuentren calificados
para ese tamaño de lote y que sean idénticos en capacidad y principio de
funcionamiento a los que se emplearan para producir los lotes a comercializar.
-4132/12
Exigencia de demostración
de bioequivalencia establecida en la Disposición ANMAT N° 3185/99, a todas las
concentraciones comercializadas y/o a comercializarse de una especialidad
medicinal, de forma farmacéutica sólida oral, que contenga alguno de los
Ingredientes Farmacéuticos Activos incluidos en la normativa nacional sobre
estudios de bioequivalencia y en disposiciones complementarias posteriores de
exigencia de bioequivalencia.
-4326/12
Adóptanse como criterios
de riesgo sanitario, para la inclusión de ingredientes farmacéuticos activos en
el cronograma de exigencia de estudios de bioequivalencia in-vivo, a los
obrantes en el Anexo I de la presente disposición.
-4788/12
Incorpóranse a las
exigencias de realización de estudios de Bioequivalencia/Biodisponibilidad,
establecidas por Disposición (ANMAT) N° 3185/99, a los ingredientes
farmacéuticos activos que figuran en el Anexo I de la presente disposición, que
forma parte integrante de la misma.
-1918/13
Establécense los criterios
para la selección de una especialidad medicinal como producto de referencia
para los estudios de Bioequivalencia y Equivalencia In-Vitro que figuran en el
Anexo I de la presente disposición, que forma parte integrante de la misma.
-2434/13
Establécese que los
titulares de laboratorios de especialidades medicinales que contengan IFAS,
respecto de los cuales esta Administración Nacional exige la realización de
estudios de Bioequivalencia, que soliciten la aprobación de resultados de
estudios realizados en el exterior, deberán cumplir con los requisitos que se
detallan en el Anexo I de la presente Disposición.
9.
REFERENCIAS
1-
Amidon G. L., Lennernäs H., Shah V. P. and Crison J. R. A. Theoretical Basis
for a Biopharmaceutics Drug Classification: The Correlation of In vitro Drug
Product Dissolution and In vivo Bioavailability. Pharmaceutical
Research,12(3):413-420, 1995.
2-
FDA, 1997. Guidance for Industry: Dissolution Testing of Immediate Release
Solid Oral Dosage Forms, US Center for Drug Evaluation and Research, USA.
3-
Fan de Waterbeend H. Fundamental Variables of the Biopharmaceutics
Classification System (BCS: A Commentary). European Journal of Pharmaceutical
Sciences, Vol 7: 1-3, 1998.
4-
Chenga C-L., Yub L. X., Leec H-L., Yangd C-Y., Luee C-S., Chen C-H. Biowaiver
extension potential to BCS Class III high solubility-low permeability drugs:
bridging evidence for metformin immediate-release tablet. European Journal
Pharmaceutical Scinces,22, 297-304, 2004.
5-
Lindenberg M., Koop S. and Dressman J.B. Classification of. orally administered
drugs on the World Health Organization Model list of Essential Medicines
according to the biopharmaceutics classification system. Eur. J. Pharm.
Biopharm., 58, 265 - 278, 2004.
6-
Polli J. E., Yu L. X., Cook J. A., Amidon G. L., Borchardt R. T., Burnside B.
A., Burton P. S., Chen M. L. Conner D. P., Faustino P. J., Hawi A. A., Hussain
A. S. Joshi H. N., Kwei Lee V. H. Lesko L. J., Lipper R. A., Loper A. E.,
Nerurkar S., Polli J. W., Sanvordeker D. R., Taneja R. Uppoor R. S., Vattikonda
C. S., Wilding I., Zhang G. Summary Workshop Report: Biopharmaceutic
Classification System-Implementation Challenges and Extension Opportunities. J.
Pharm. Sci., 93 (6), 1375 - 1381, 2004.
7-
FDA, 2000. Guidance for industry: Waiver of In vivo Bioavailability and
Bioequivalence Studies for Immediate Release Solid Oral Dosage Forms based on
Biopharmaceutics Classification System. US Food and Drug Administration, Center
for Drug Evaluation and Research, USA.
8-
Yu L.X., Amidon G., Polli J., Zhao H., Mehta M., Conner D., Shah V., Lesko L.,
Chen M, Lee V. and Hussain A. Biopharmaceutics Classification System: The
Scientific Basis for Biowaiver Extensions, Pharml Res., 19(7):921-5, 2002.
9- Bermejo M. and Amidon G.
“Bioequivalencia In Vitro. ¿Porqué, cuando y cómo Aplicación del BCS (Sistema
de Clasificación Biofarmacéutico). En: Primer Encuentro Iberolatinoamericano de
Academias de Farmacia. Valparaíso Chile, Abril 2005.
10-
Yazdanian N., Briggs K., Jankosky C. and Hawi A. The “High Solubility”
definition on the current FDA Guidance on Biopharmaceutical Classification
System may be too strict for acidic drugs. Pharml Res, 21(2): 293-299, 2004.
11-
Le Cluyse E. and Sutton, S. In vitro models for selection of development
candidates. Permeability Studies to defined mechanisms of absorption
enhancement. Adv. Drug Del. Rey., 23:163-183, 1997.
12- Farmacopea Nacional
Argentina. Séptima edición.
13-
USP 38. United States Pharmacopeia
14-
FDA, 1987. Guideline on General Principles of Process Validation. US Food and
Drug Administration, Center for Drug Evaluation and Research, USA.
15- ANVISA. Dispõe sobre o
Guia para isençáo e substituição de estudos de biodisponibilidade
relativa/bioequivalência e dá outras providências. RESOLUÇÃO. RDC N° 37, 3 DE
AGOSTO DE 2011.
16- Instituto de Salud
Pública de Chile, 2007. Guía Técnica G-BIOF 02: Bioexención de los estudios de
Biodisponibilidad/Bioequivalencia para establecer Equivalencia Terapéutica de
Formas Farmacéuticas Sólidas Orales. Sección de Biofarmacia.
17- FDA, 2000. Formas
posológicas orales sólidas de liberación inmediata Cambios de escala y
posteriores a la aprobación: documentación química, de fabricación y controles,
de pruebas de disolución in vitro y bioequivalencia in vivo. US
Food and Drug Administration, Center for Drug Evaluation and Research, USA.
18-
Health Canada. Release of Guidance Document: Biopharmaceutics Classification
System Based Biowaiver. May 30, 2014; File number: 14-105447-315.
19-
European Medicines Agency (EMA), Guideline on the Investigation of
Bioequivalence. London, 20 January 2010.
20-
WHO technical report series. WHO Expert Committee on Specifications for
PharmaceuticaI Preparations. Forty-nineth report. Geneva, World Health
Organization, 2015 (WHO Technical Report Series, No. 992): 131.
DOCUMENTACIÓN REQUERIDA
PARA LA SOLICITUD DE BIOEXENCIÓN SEGÚN DISPOSICIÓN ANMAT N° 758/09
Anexo I: Bioexención
basada en la Clasificación Biofarmacéutica.
SOLICITANTE:
OBJETIVO DEL ESTUDIO:
Índice de la Presentación
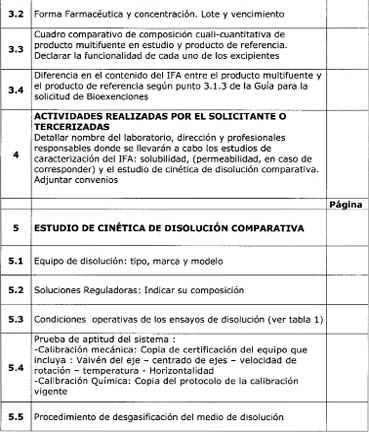



Tabla 1: Datos de
disolución in vitro para la Bioexención requerida.
|
Condiciones de disolución
|
|
|
Equipo
|
|
|
RPM
|
|
|
Medio
|
|
|
Volumen
|
|
|
Temperatura
|
|
|
Enzimas
|
|
|
Volumen de muestra
|
|
DOCUMENTACIÓN REQUERIDA
PARA LA SOLICITUD DE BIOEXENCIÓN SEGÚN DISPOSICIÓN ANMAT N° 758/09
Anexo II: Bioexenciones
basadas en formulaciones proporcionalmente similares.
SOLICITANTE:
OBJETIVO DEL ESTUDIO:
Índice de la Presentación
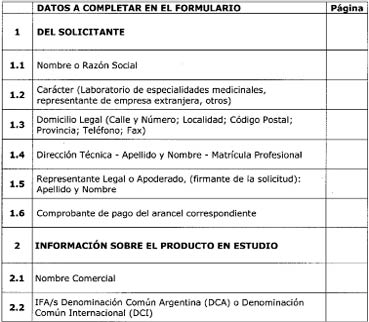
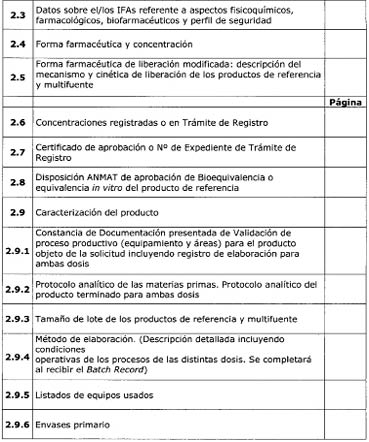



Tabla 1: Datos de
disolución in vitro para la Bioexención requerida
|
Condiciones de
disolución
|
|
|
Equipo
|
|
|
RPM
|
|
|
Medio
|
|
|
Volumen
|
|
|
Temperatura
|
|
|
Enzimas
|
|
|
Volumen de muestra
|
|
DOCUMENTACIÓN REQUERIDA
PARA LA SOLICITUD DE BIOEXENCIÓN SEGÚN DISPOSICIÓN ANMAT N° 556/09
Anexo III: Cambios
posteriores a la aprobación del registro de un producto farmacéutico que ha
demostrado bioequivalencia o equivalencia in vitro basado en la clasificación
biofarmacéutica.
SOLICITANTE:
OBJETIVO DEL ESTUDIO:
Índice de la Presentación
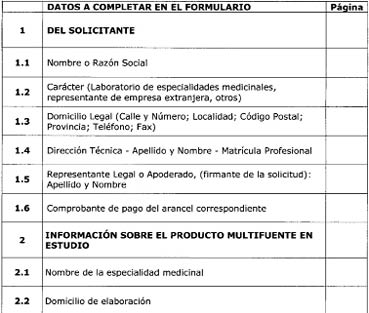
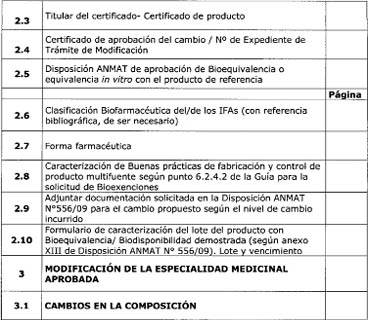

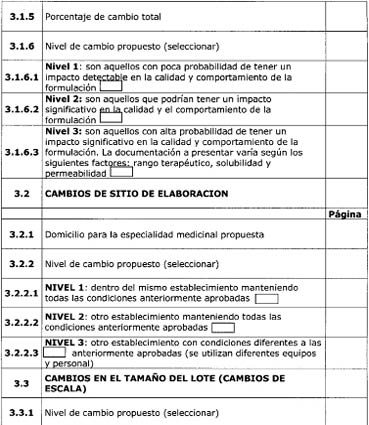
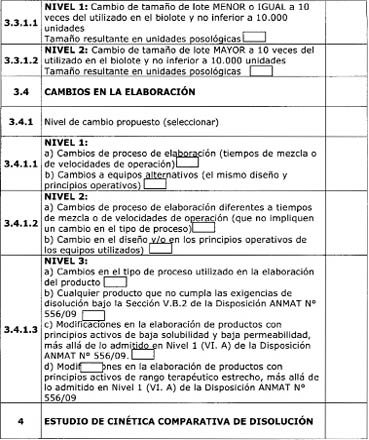
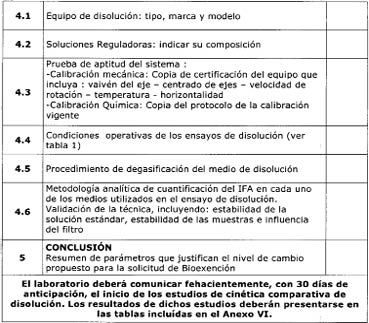
Tabla 1: Datos de
disolución in vitro para la Bioexención requerida.
Condiciones de disolución
Equipo
RPM
Medio
Volumen
Temperatura
Enzimas
Volumen de muestra
DOCUMENTACIÓN REQUERIDA
PARA LA PRESENTACIÓN DE RESULTADOS PARA SOLICITAR UNA BIOEXENCIÓN SEGÚN
DISPOSICIÓN ANMAT N° 758/09.
Anexo IV: Bioexención
basada en la Clasificación Biofarmacéutica
INFORMACIÓN – DOCUMENTO
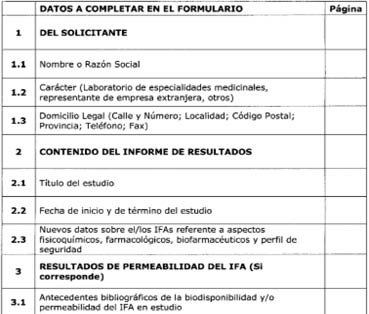


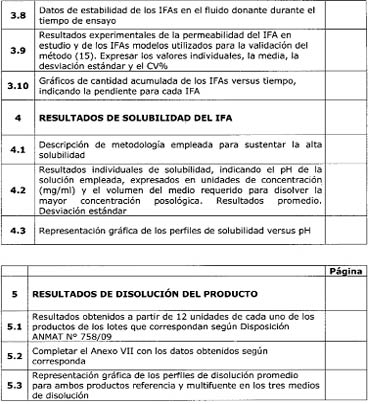
DOCUMENTACIÓN REQUERIDA
PARA LA PRESENTACIÓN DE RESULTADOS DE PROPORCIONALIDAD DE DOSIS PARA SOLICITAR
UNA BIOEXENCIÓN SEGÚN DISPOSICIÓN ANMAT N° 758/09
Anexo V: Bioexenciones
basadas en formulaciones proporcionalmente similares
INFORMACIÓN – DOCUMENTO


DOCUMENTACIÓN REQUERIDA
PARA LA PRESENTACIÓN DE RESULTADOS DE CAMBIOS POSTERIORES A LA APROBACIÓN DE UN
REGISTRO PARA SOLICITAR UNA BIOEXENCIÓN SEGÚN DISPOSICIÓN ANMAT N° 556/09
Anexo VI: Cambios
posteriores a la aprobación del registro de un producto farmacéutico que ha
demostrado Bioequivalencia o equivalencia in vitro
INFORMACIÓN – DOCUMENTO


DOCUMENTACIÓN REQUERIDA
PARA LA PRESENTACIÓN DE RESULTADOS DE ESTUDIOS DE CINETICA DE DISOLUCIÓN
Anexo VII: Planillas para
la presentación de los resultados individuales de los estudios de cinética de
disolución.
Planillas modelo para
completar con los resultados de los estudios cinéticos para ambos productos en
cada uno de los medios requeridos. Se deberá presentar una planilla para cada
producto en cada medio utilizado por separado.
Las planillas contienen
datos mínimos a ser presentados de los estudios cinéticos y deberán estar
acompañadas de los datos crudos. Asimismo cualquier información adicional podrá
ser adjuntada en este anexo.