Disposición 758-2009
Criterios de Bioexención de Estudios de
Bioequivalencia para medicamentos sólidos orales de liberación inmediata.
Bs. As., 23/2/2009
VISTO la Disposición (ANMAT) Nº 3185/99, el
Informe de OMS 937/2006 (Anexo 8), y el Expediente Nº 1-47-1110-749-08-1 del
Registro de la Administración Nacional de Medicamentos Alimentos y Tecnología
Médica, y;
CONSIDERANDO:
Que los estudios de bioequivalencia en
humanos han sido aceptados en los últimos 25 años como un requisito sobre el
cual se basan las agencias regulatorias de medicamentos para establecer la
equivalencia terapéutica de los productos farmacéuticos multifuentes con
respecto a un producto de referencia.
Que por Disposición (ANMAT) Nº 3185/99 se
aprobaron recomendaciones para la realización de estudios de bioequivalencia
entre medicamentos con riesgo sanitario significativo, y se estableció un
cronograma de implementación gradual en consideración a los antecedentes
internacionales en la materia, y los requerimientos in vivo e in vitro
requeridos para los productos allí clasificados.
Que la Organización Mundial de la Salud (OMS)
en su Serie de Informes Técnicos Nº 937 —Anexo 8— ha considerado al Sistema de
Clasificación Biofarmacéutica (SCB), como una herramienta válida de
clasificación de aquellos principios activos que solo requieren demostraciones
de equivalencia in vitro exceptuándolos de estudios in vivo.
Que el SCB se basa en la solubilidad acuosa y
la permeabilidad intestinal del principio activo y cuando estas dos
propiedades, se combinan con la disolución del medicamento se obtienen los tres
factores que determinan la velocidad y la cantidad de principio activo
absorbido desde una forma farmacéutica oral de liberación inmediata,
permitiendo establecer la inferencia de la Bioequivalencia.
Que dicha inferencia entre dos productos
medicinales de acuerdo al SCB se basa en que: Si dos productos tienen el mismo
perfil disolución in vivo a lo largo del tránsito intestinal tendrán el mismo
perfil de concentración / tiempo en la superficie de la membrana intestinal lo
que implica que ambos productos tendrán una similar velocidad de absorción del
principio activo y por lo tanto similar biodisponibilidad.
Que de acuerdo a los principios del SCB
surgen dos conceptos aplicables a los productos farmacéuticos multifuentes: el
de estudios de bioequivalencia in vitro y el de bioexenciones.
Que los estudios de bioequivalencia in vitro
se refieren a aquellas pruebas de disolución in vitro que determinan la
similaridad de los perfiles de disolución entre el producto multifuente y el
producto comparador, mientras que el término bioexención se aplica en la
práctica regulatoria cuando un producto farmacéutico se aprueba en base a
evidencias de equivalencia que no involucran pruebas de bioequivalencia in
vivo.
Que la introducción del SCB ha provocado un
gran impacto en la práctica regulatoria al establecer las pautas para la
demostración de la bioequivalencia entre medicamentos mediante ensayos de
disolución in-vitro para las formas farmacéuticas sólidas orales de liberación
inmediata, limitando así los requerimientos de estudios in vivo.
Que las agencias de medicamentos de los
Estados Unidos (Food and Drug Administration) y de la Comunidad Económica
Europea (European Medicines Agency) han adoptado los criterios del SCB y ambas
coinciden en que la bioexención es justificada cuando las drogas tienen alta
solubilidad y alta permeabilidad (Clase I), son de amplio margen terapéutico y
el producto cumple con determinados criterios de disolución.
Que la OMS en el documento antedicho
considera bioexceptuables a las drogas de Clase I y también extiende la
bioexención a los productos medicinales que contengan drogas de Clase III (baja
permeabilidad / alta solubilidad).
Que en nuestro país ya existen antecedentes
de la aplicación de los criterios del SCB, así la Disposición (ANMAT) Nº
3311/01, considera bioexceptuables los productos medicinales conteniendo
ciertas drogas antirretrovirales.
Que la demostración de la bioequivalencia
mediante ensayos de disolución, permite reducir en forma considerable los
tiempos y costos de la realización de estudios in vivo, reemplazar en algunos
casos los estudios en humanos y agilizar la ejecución de las políticas
sanitarias vigentes con la finalidad de poner al alcance de la población un
número cada vez mayor de productos medicinales con equivalencia establecida.
Que la Disposición (ANMAT) Nº 3185/99 refiere
a las recomendaciones técnicas para la realización de los estudios de
bioequivalencia e incluye un cronograma de exigencias de estudios in vivo para
drogas de estrecho margen terapéutico e incorpora además un listado de drogas
confeccionado de acuerdo a la Lista de Medicamentos Esenciales de la OMS los
cuales fueron caracterizados de acuerdo a su riesgo sanitario en alto,
intermedio y bajo.
Que las drogas incluidas dentro de la
categoría de riesgo intermedio y bajo poseen amplio rango terapéutico y un
número importante de ellas pertenece a las Clases I y III por lo que los
productos medicinales correspondientes pueden ser considerados bioexceptuables
de acuerdo a lo recomendado en los documentos de la OMS.
Que el Instituto Nacional de Medicamentos ha
evaluado los principios activos denominados de Riesgo Intermedio y Bajo según
la Disposición Nº 3185/99 sobre la base de los criterios del SCB, y ha
efectuado la propuesta de los que deberían incluirse en la presente Normativa
de Bioexención.
Que la Comisión Asesora Ad Honórem de la
ANMAT en Temas de Bioequivalencia y Biodisponibilidad y la Dirección de Asuntos
Jurídicos han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades
conferidas por el Decreto Nº 1490/92 y Decreto Nº 253/08.
Por ello,
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL
DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE
Artículo 1º — Adóptanse los Criterios de Bioexención de Estudios
de Bioequivalencia, basados en la Clasificación Biofarmacéutica para las formas
farmacéuticas sólidas orales de liberación inmediata, con alcance para algunos
de los principios activos establecidos en la Disposición (ANMAT) Nº 3185/99
como de riesgo intermedio y los que en un futuro se vayan incorporando, así
como las exigencias que se deberán cumplir para demostración de la Bioexención,
los cuales figuran como Anexo I de la presente Disposición.
Art. 2º — Establécese que los criterios adoptados en el artículo 1º serán de
aplicación para todos los laboratorios poseedores de certificados de
medicamentos conteniendo los principios activos de riesgo intermedio alcanzados
por la presente normativa.
Art. 3º — Será requisito previo para la inscripción en el Registro de
Especialidades Medicinales de aquellos productos que contengan alguno de los
principios activos a los que se hace referencia en el art. 1º de la presente
(artículos 3º, 4º y 5º del Decreto 150/92), la realización de los estudios que
sustenten la bioexención según los criterios y condiciones establecidos en la
presente norma.
Art. 4º — No se autorizará la comercialización, bajo cualquier modalidad, de
aquellas especialidades medicinales que contengan alguno de los principios
activos a los que hace referencia el art. 1º de la presente Disposición y que a
la fecha no se encuentren comercializadas, hasta tanto cumplan con la
realización de los estudios de equivalencia, de conformidad con lo establecido
con la presente Disposición.
Art. 5º — La falta de demostración satisfactoria de los criterios de
bioexención enunciados en el ANEXO I de la presente Disposición producirá la
denegatoria del pedido de bioexención, para la formulación presentada.
Art. 6º — Esta Administración Nacional de Medicamentos, Alimentos y Tecnología
Médica, dentro del plazo de ciento ochenta (180) días corridos, contados a
partir del día siguiente al de su publicación en el Boletín Oficial, dictará
las normas complementarias mediante las cuales establecerá los lineamientos
operativos para la aplicación efectiva de la presente Disposición.
Art. 7º — La presente Disposición entrará en vigencia a partir del día
siguiente del término del plazo establecido en el artículo anterior.
Art. 8º — Regístrese. Dése a la Dirección Nacional del Registro Oficial para su
publicación. Notifíquese a las Cámaras de Especialidades Medicinales (CILFA,
CAEME, COOPERALA, CAPGEN, CAPEMVeL, SAFYBI), Confederación Médica de la
República Argentina (COMBA) y la Confederación Farmacéutica Argentina (COFA)
Cumplido, archívese PERMANENTE. — Ricardo Martínez.
ANEXO I
CRITERIOS
DE BIOEXENCION DE ESTUDIOS DE BIOEQUIVALENCIA PARA MEDICAMENTOS SOLIDOS ORALES
DE LIBERACION INMEDIATA.
I) BIOEXENCION BASADA EN LA CLASIFICACION
BIOFARMACEUTICA.
En el marco de la Clasificación
Biofarmacéutica de los principios activos y tomando como base las propiedades
de disolución de la forma farmacéutica, las especialidades medicinales
formuladas con drogas de Riesgo intermedio, podrán ser exceptuadas de la
obligatoriedad de realizar estudios de bioequivalencia.
Para ser exceptuado de un estudio de
bioequivalencia in vivo, un medicamento oral sólido de liberación inmediata
deberá demostrar características de disolución muy rápida o rápida, dependiendo
de las propiedades del principio activo en términos de la Clasificación
Biofarmacéutica.
Los resultados de perfiles de Disolución
deberán asimismo demostrar similaridad con los correspondientes al Producto
Comparador de Referencia.
Los excipientes incluidos en la composición
de la forma farmacéutica, deberán ser considerados conforme a lo indicado a
continuación:
SISTEMA DE CLASIFICACION BIOFARMACEUTICA
Es un marco científico para clasificar
principios activos sobre la base de su solubilidad acuosa y su permeabilidad
intestinal. Cuando se combinan con la disolución del medicamento, el SCB toma
en cuenta estos tres factores: disolución, solubilidad y permeabilidad
intestinal que gobiernan la velocidad y la cantidad de absorción (exposición)
de principio activo liberado desde una forma farmacéutica sólida oral de
liberación inmediata
Clasificación de las drogas:
Clase I: Alta Solubilidad - Alta
permeabilidad
Clase II: Baja Solubilidad - Alta
permeabilidad.
Clase III: Alta Solubilidad - Baja permeabilidad
Clase IV: Baja Solubilidad - Baja
permeabilidad
Definiciones:
250 ml en el rango de pH 1.2-6.8 a 37º C. =Alta
Solubilidad: La dosis más alta es soluble en un volumen
Alta Permeabilidad: Más del 85% de la dosis
oral administrada se absorbe en el intestino delgado.
Disolución Muy Rápida: Más del 85% de la
cantidad declarada se disuelve dentro de los 15 min. en medio estándar a pH
1.2, 4.5 y 6.8 usando el aparato II a 75 rpm o alternativamente el aparato I a
100 rpm.
Disolución Rápida: Más del 85% de la cantidad
declarada se disuelve dentro de los 30 min. en medio estándar a pH 1.2, 4.5 y
6.8 usando el aparato II a 75 rpm o alternativamente el aparato I a 100 rpm.
II) BIOEXENCION BASADA EN FORMULACIONES
PROPORCIONALMENTE
SIMILARES.
Cuando la dosis más alta de un producto
multifuente hubiere demostrado equivalencia in vivo o in vitro con el producto
comparador de referencia según se estableció en las normativas de
Bioequivalencia precedentes, o en la presente Normativa, los productos de menor
dosis no requerirán estudios comparativos con el producto comparador de
referencia, si cumplen con las siguientes condiciones:
1) La composición de las distintas dosis es
proporcionalmente similar al producto originalmente bioexceptuado.
2) Los perfiles de disolución demuestren ser
similares entre las distintas dosis. Dos formulaciones se consideran
proporcionalmente similares si:
a) Todos los ingredientes activos e inactivos
de dos dosis distintas, están en la misma proporción.
b) Todos los ingredientes inactivos de dos
dosis distintas son los mismos y se encuentran en la misma cantidad y el peso
de la forma farmacéutica total es casi el mismo.
III) DROGAS DE RIESGO INTERMEDIO.
|
Propranolol
|
Salbutamol
|
|
Tamozifeno
|
Amitriptilina
|
|
Diazepan
|
Atenolol
|
|
Etambutol
|
|
|
Hidralazina
|
Flucitosina
|
|
Metildopa
|
Clormiprámina
|
|
Biperideno
|
Quinina bisulfato
|
IV) SELECCION DE LOS PRODUCTOS COMPARADORES
DE REFERENCIA:
(Punto derogado por art. 4° de la Disposición N° 1918/2013 de la
Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O.
15/4/2013. Vigencia: a partir del primer día hábil siguiente al de su
publicación en el Boletín Oficial)
V) EXCIPIENTES:
Se deberá establecer la adecuabilidad de los
excipientes aportando documentación correspondiente a cualquiera de las
siguientes alternativas:
1- El/los excipiente/s están presentes en
cantidades similares en el Producto Comparador de Referencia.
2- El /los excipientes están presentes en
cantidades similares en productos que contiene el mismo principio activo y
poseen autorización de comercialización en países miembros de ICH.
3 - El /los excipientes están presentes en el
producto en cantidades normalmente empleadas para esa forma farmacéutica, para
documentar lo cual puede consultarse los "sites":
www.fda.gov/cder/iig/iigfaqWEB.htm
www.accessdata.fda.gov/scripts/cder/iig/index.cfm
VI) EXIGENCIAS DE COMPORTAMIENTO DE
DISOLUCION PARA LA BIOEXENCION DE PRODUCTOS FARMACEUTICOS.
Drogas de Clase I
a) Presentan rápida disolución (85% de la
cantidad declarada de droga se disuelve en 30 min. o menos) y el perfil de
disolución del producto multifuente es similar al Producto Comparador de
Referencia (f2 > 50) en los tres medios: pH: 1.2, 4.5 y 6.8 utilizando
Aparato I a 100 rpm o Aparato II a 75 rpm.
b) Tanto el producto multifuente como el
Producto Comparador de Referencia presentan muy rápida disolución (85% de la
cantidad de declarada de droga se disuelve en 15 min. o menos) en los tres
medios antes mencionados. En este caso, no es necesaria la determinación de los
perfiles de disolución.
Drogas de Clase III
a) Tanto el Producto Comparador de Referencia
como el producto multifuente presentan muy rápida disolución (85% o más de la
cantidad declarada de droga se disuelve en 15 min. o menos) a pH 1.2, 4.5 y 6.8
utilizando Aparato I a 100 rpm o Aparato II a 75 rpm.
3) Condiciones operativas para la obtención
de Perfiles de Disolución comparativos. Las pruebas de disolución se realizarán
en Aparato I a 100 rpm o Aparato II a 75 rpm (Farmacopea Argentina, 7º Ed.
Vol.1) usando 900 ml o menos de los siguientes medios de disolución: solución
HC1 pH 1.2, buffer acetato pH 4.5 y buffer fosfato pH 6.8 o fluido intestinal
simulado sin enzimas, a 37º C.
El Aparato I (canastilla) se prefiere en
general para cápsulas y productos que floten, y el Aparato II (paletas) se
prefiere en general para comprimidos.
Para cápsulas de gelatina puede ser usado
fluido gástrico simulado con enzimas. Deberá ser evaluado un mínimo de 12
unidades del producto.
Los intervalos de toma de muestra deberán ser
suficientes como para poder caracterizar completamente el perfil de disolución,
por ej.: 10, 15, 20, 30, 45 y 60 minutos). La comparación de los perfiles de
disolución entre productos se determina mediante el Factor de similaridad (f2).
Un valor f2 de 50 o mayor (50-100) asegura similitud o equivalencia entre las
dos curvas.
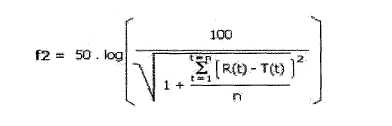
Donde R y T corresponden al porcentaje
acumulado de droga disuelta del Producto Comparador de Referencia (R) y del
producto en estudio (T) respectivamente a cada intervalo de tiempo n: Número de
tomas de muestra durante el ensayo de disolución. Para su cálculo deben
cumplirse las siguientes condiciones: Se dispone como mínimo de tres tiempos de
muestreo , el coeficiente de variación debe ser inferior al 20% en los primeros
tiempos e inferior al 10% en los últimos tiempos, los tiempos de toma de
muestra deben ser los mismos para ambas formulaciones. Un solo tiempo de
muestreo es suficiente luego de que el producto comparador alcanzó el 85% de
disolución. Cuando el 85% de la concentración declarada del producto se
disuelve en 15 min. usando los tres medios recomendados, no es necesario
realizar la comparación f2.