Disposición
2372-2008
Apruébanse
la "Guía para Inspectores sobre Buenas Prácticas de Fabricación de
Medicamentos’’ y la "Clasificación de Deficiencias de Cumplimiento de las
Buenas Prácticas de Fabricación’’.
Bs. As., 24/4/2008
VISTO el
Expediente Nº 1-47-1110-537-07-5 del Registro de esta Administración Nacional
de Medicamentos, Alimentos y Tecnología Médica; y
CONSIDERANDO:
Que de acuerdo con
su decreto de creación 1490/92, esta Administración Nacional de Medicamentos,
Alimentos y Tecnología Médica tiene competencia en todo lo referido al control
y fiscalización sobre la sanidad y calidad, entre otros productos, de las especialidades
medicinales y al contralor de las actividades, procesos y tecnologías que se
realicen en función del aprovisionamiento, producción, elaboración,
fraccionamiento, importación y/o exportación, depósito y comercialización de
los productos, sustancias, elementos y materiales consumidos o utilizados en la
medicina humana (Artículo 3 incisos a) y e) del aludido decreto).
Que en el marco de
la referida competencia, el Decreto 1490/92 confiere a esta Administración
Nacional atribuciones para autorizar y registrar los referidos productos,
fiscalizar el cumplimiento de las normas de sanidad y calidad establecidas para
éstos así como también proceder a la habilitación de las personas físicas o
jurídicas que intervengan en las acciones de aprovisionamiento, producción,
elaboración, fraccionamiento, importación y/o exportación, depósito,
comercialización de especialidades medicinales.
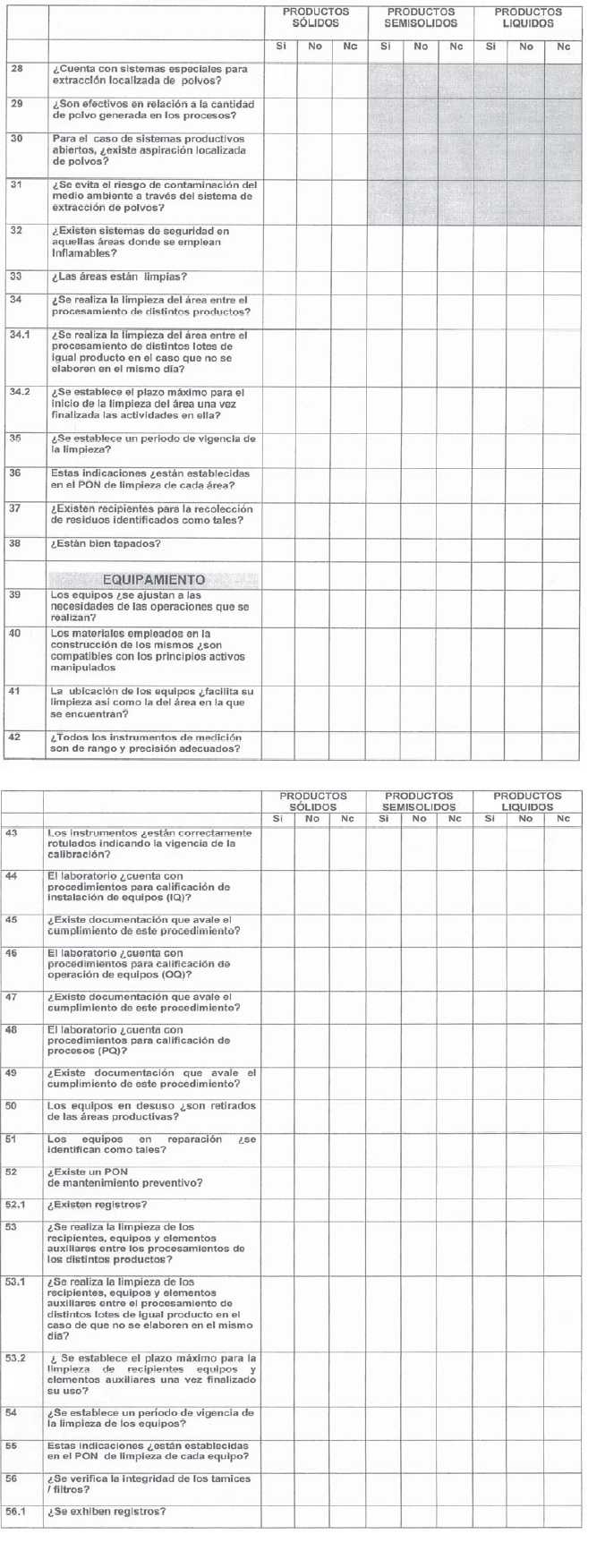
Que tales
actividades tienen como objetivo primordial garantizar a la población la
eficacia, seguridad y calidad de los productos que consume.
Que uno de los
mecanismos idóneos que contribuyen a garantizar la calidad con que llegan al
mercado los productos que elaboran, importan y distribuyen los establecimientos
productores, importadores y distribuidores de especialidades medicinales es la
fiscalización y control de tales establecimientos a través de inspecciones
técnicas que cubran aspectos relativos a las condiciones de funcionamiento y
sistemas de control de calidad utilizados en los establecimientos en cuestión.
Que con el fin de
llevar cabo estas acciones de control, previstas en el artículo 8 inciso n) del
Decreto 1490/92, resulta necesario contar con un modelo que asegure el control
de las industrias con uniformidad de criterio así como también la neutralidad,
simetría y reciprocidad en el tratamiento y aplicación de las normas
regulatorias.
Que en ese
entendimiento se dictó la Disposición ANMAT Nº 2819/04, que adoptó las
recomendaciones vigentes sobre Buenas Prácticas de Fabricación para la Industria Farmacéutica aprobadas por la Asamblea Mundial de la Salud; y los informes de la PIC’S (Pharmaceutical Inspection Cooperation Scheme) y de la ICH (International Conference on Harmonisation), cuyo cumplimiento, o el de la norma que en
el futuro pudiera reemplazarla, es obligatorio para los establecimientos
productores, importadores y distribuidores de Especialidades Medicinales.
Que dentro del
concepto de Garantía de Calidad, las Buenas Prácticas de Fabricación
constituyen el factor que asegura que los productos se fabriquen en forma uniforme
y controlada de acuerdo con las normas de calidad adecuadas al uso que se
pretende dar a los productos y conforme a las condiciones exigidas para su
comercialización.
Que teniendo en
cuenta la conveniencia de capacitar en forma continua a los inspectores y con
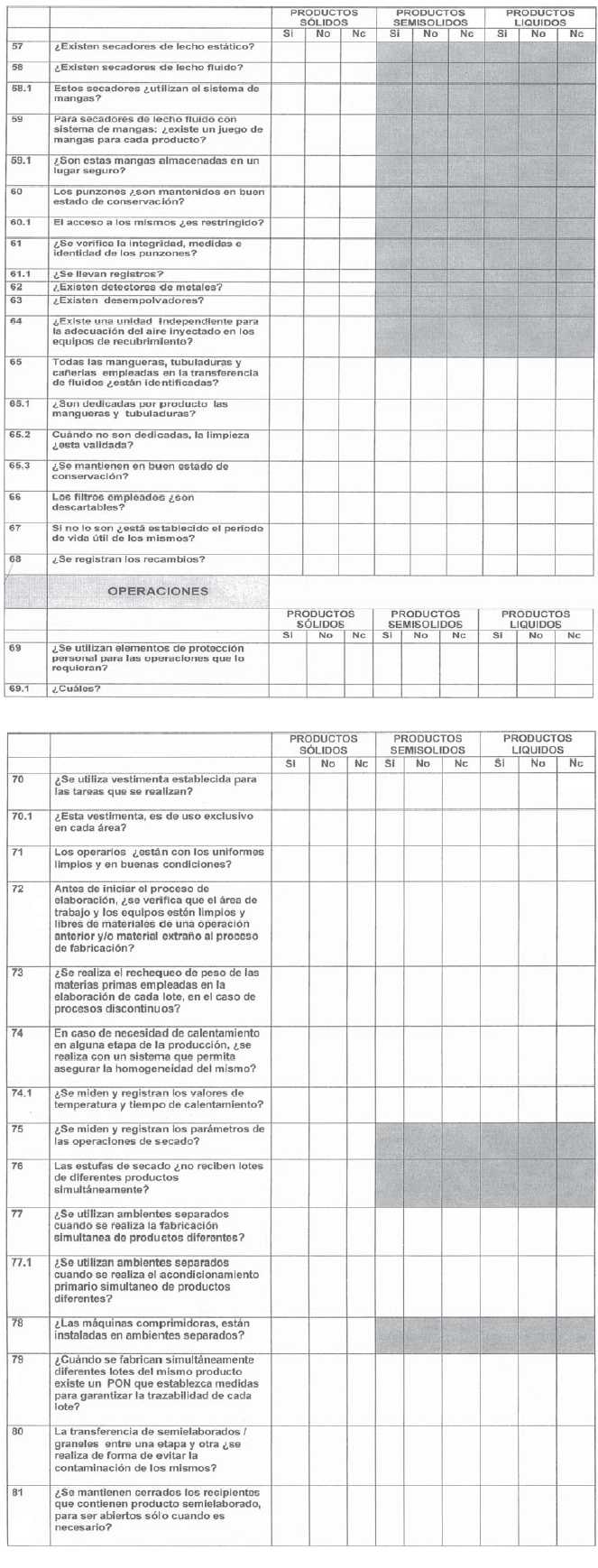
el objeto de servir como documento de apoyo tanto para la autoridad reguladora
en las inspecciones como para la industria en la verificación y aplicación de
las normas de buenas Prácticas de fabricación farmacéuticas resulta conveniente
aprobar una "Guía para Inspectores sobre Buenas Prácticas de Fabricación
de Medicamentos", teniendo en cuenta los lineamientos de la Disposición ANMAT Nº 2819/04 o de la que en el futuro la reemplace.
Que por otra parte
el objetivo de las inspecciones que lleva a cabo esta Administración Nacional
no es sólo verificar la adecuación de la empresa a las normas de buenas
prácticas de fabricación sino también orientarla en la modificación de
procedimientos de producción, distribución y comercialización que pueden resultar
riesgosos para la salud, promoviendo a través de estas dos tareas principales
un impacto positivo en el aseguramiento de la calidad de los medicamentos.
Que en su rol de
verificadora de las normas de BPF esta Administración advierte la existencia de
deficiencias de cumplimiento de la BPF y en uso de las facultades conferidas
por el Artículo 8º inciso ñ) del Decreto 1490/92 y el Decreto 341/92 puede
adoptar las medidas más oportunas y adecuadas para proteger la salud de la
población, conforme a la normativa vigente.
Que las referidas
deficiencias pueden implicar consecuencias de diversa envergadura para las
empresas involucradas, por lo cual y con el objeto de asegurar la uniformidad
de criterios en la aplicación de la normativa, resulta conveniente que tales
deficiencias sean claramente determinadas, no sean ambiguas y se basen en las
regulaciones aplicables.
Que como
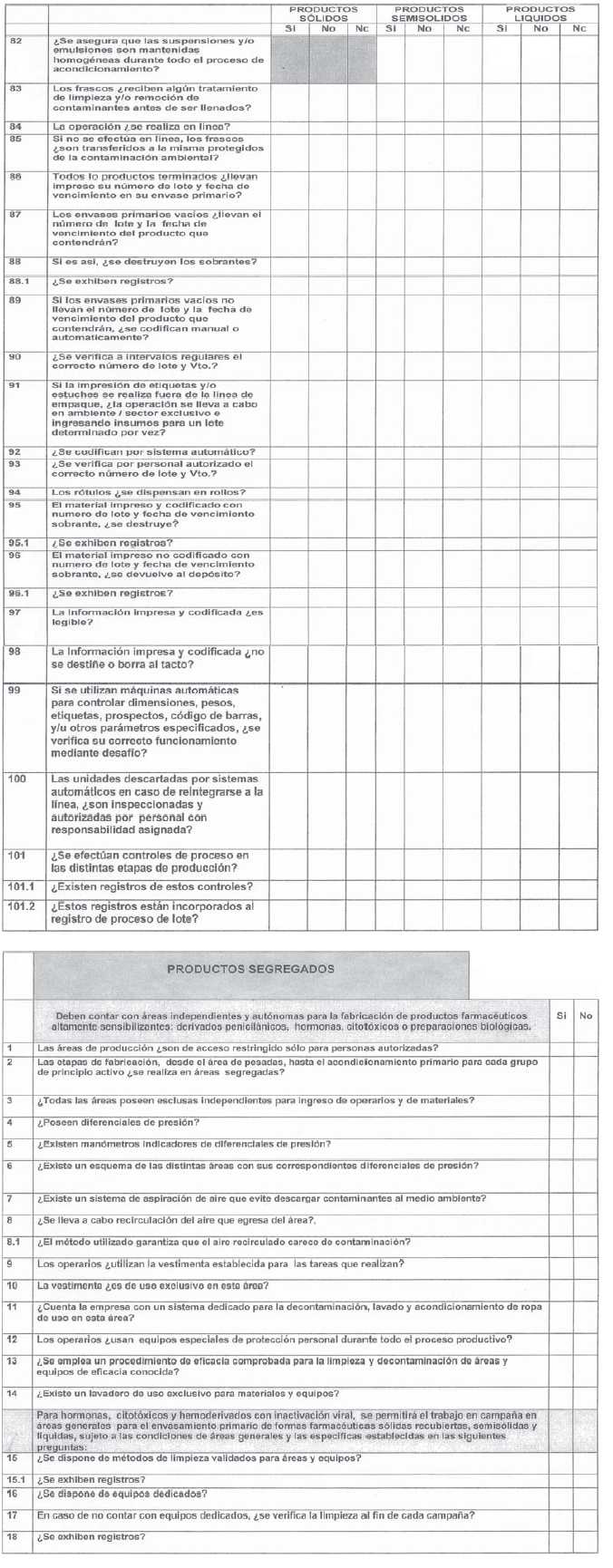
consecuencia de ello es indispensable proceder a la clasificación de las
deficiencias de cumplimiento de las normas de Buenas Prácticas de Fabricación
en deficiencias críticas, mayores y otras deficiencias, adoptándose, a esos
fines, un criterio basado en el riesgo involucrado teniendo en cuenta la
naturaleza y el alcance de deficiencia advertida.
Que finalmente y
con el objeto de garantizar la transparencia y equidad en los procedimientos
seguidos y la proporcionalidad de las acciones a adoptar resulta conveniente
relacionar cada deficiencia de cumplimiento con la medida de prevención que
corresponderá implementar en cada caso.
Que a los efectos
de la presente han sido considerados como antecedentes los documentos de la
agencia sanitaria de Canadá.
Que el Instituto
Nacional de Medicamentos y la Dirección de Asuntos jurídicos han tomado la
intervención de su competencia.
Que se actúa en
virtud de las facultades conferidas por el Decreto Nº 1490/92 y el Decreto Nº
253/08.
Por ello;
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1º — Apruébase la "Guía para Inspectores sobre
Buenas Prácticas de Fabricación de Medicamentos", que como Anexo I forma
parte integrante de la presente disposición.
Art. 2º — Apruébase la "Clasificación de Deficiencias de
Cumplimiento de las Buenas Prácticas de Fabricación", que como Anexo II
forma parte integrante de la presente disposición.
Art. 3º — Establécese que la presente disposición entrará en
vigencia a partir del día siguiente al de su publicarán en el Boletín Oficial.
Art. 4º — Derógase el artículo 2º de la Disposición ANMAT Nº 1930/95, la Disposición ANMAT Nº 1231/94, la Disposición ANMAT Nº 853/99 y la Disposición ANMAT Nº 2672/99.
Art. 5º — Anótese; comuníquese a CAEME, COOPERALA, CILFA,
CAPGEN y CAPDROFAR. Dése a la Dirección Nacional del Registro Oficial para su publicación. Cumplido, archívese PERMANENTE. — Ricardo Martínez.
ANEXO I


















ANEXO II
|
CLASIFICACION DE DEFICIENCIAS
DE CUMPLIMIENTO DE LAS
BUENAS PRACTICAS DE FABRICACION
|
v Medidas a tomar
sobre la base de las deficiencias observadas:
v CLAUSURA
v CLAUSURA Y
RETIRO DEL MERCADO
v INHIBICION
v INHIBICION Y
RETIRO DEL MERCADO
v INHIBICION DE
LINEA / AREA / LOTE
v INHIBICION DE
LINEA / AREA / LOTE Y RETIRO DEL MERCADO
v RETIRO DEL
MERCADO
v MEDIDA
CORRECTIVA INMEDIATA
v MEDIDA
CORRECTIVA MEDIATA
v MEDIDA
CORRECTIVA PROGRAMADA
l Alcance de las
Medidas
v CLAUSURA: Esta
medida se aplicará cuando existan DEFICIENCIAS CRITICAS de cumplimiento de las
Buenas Prácticas de Fabricación. La clausura implicará la suspensión total o
parcial de las actividades productivas, de apoyo analítico, y de la
comercialización de los productos que se encuentren en depósito propio y/o de
terceros (distribuidoras inclusive). Puede ameritar el RETIRO DEL MERCADO del
producto, del lote involucrado o de todos los productos que comercializa el
establecimiento.
v INHIBICION: Esta
medida se aplicará cuando existan DEFICIENCIAS MAYORES de cumplimiento de las
Buenas Prácticas de Fabricación. La inhibición implicará la suspensión total o
parcial de las actividades productivas y de apoyo analítico, pudiendo ameritar
el RETIRO DEL MERCADO del producto, del lote involucrado o de todos los
productos que comercializa el establecimiento. La inhibición podrá ser de toda
la planta, o de uno o más lotes de producto, o bien de una o más líneas
productivas o de una o más áreas.
v RETIRO DEL
MERCADO: Se dispondrá teniendo en cuenta el potencial riesgo sanitario.
v MEDIDA
CORRECTIVA INMEDIATA: Son aquellas medidas correctivas de OTRAS DEFICIENCIAS de
cumplimiento de las Buenas Prácticas de Fabricación que deben implementarse en
un tiempo mínimo, durante el transcurso de la inspección.
v MEDIDA
CORRECTIVA MEDIATA: Son aquellas medidas correctivas de OTRAS DEFICIENCIAS de
cumplimiento de las Buenas Prácticas de Fabricación que deben implementarse en
un lapso de tiempo no superior a los 30 (treinta) días corridos contados a
partir del cierre del acta de inspección.
v MEDIDA
CORRECTIVA PROGRAMADA: Son aquellas medidas correctivas de OTRAS DEFICIENCIAS
de cumplimiento de las Buenas Prácticas de Fabricación, cuya implementación
requiere la presentación de un programa el cual deberá ser consensuado con la Autoridad Sanitaria.
A los fines de la
aplicación de la presente disposición se adoptan las siguientes definiciones:
DEFICIENCIA
CRITICA: Se entiende por deficiencia crítica de cumplimiento de las BPF a toda
deficiencia que pueda afectar en forma directa la calidad del producto.
DEFICIENCIA MAYOR:
Se entiende por deficiencia mayor de cumplimiento de las BPF a toda deficiencia
que, sin clasificarse como crítica, pueda derivar en un producto que no cumple
con su autorización de comercialización; o a toda deficiencia que indica una
falla en los procedimientos de liberación de lotes; o a una suma de "OTRAS
DEFICIENCIAS" que por sí solas no se clasifiquen como mayores, pero que
juntas pueden representar una deficiencia mayor.
OTRAS
DEFICIENCIAS: Deficiencias de cumplimiento de las BPF que sin clasificarse como
críticas ni como mayores, representan un desvío de dicha norma.
PLAN DE
VALIDACION: Se entenderá como tal aquel que al momento de la inspección se
encuentre en curso, de acuerdo a su programa y con un mínimo de 6 (seis) meses
de comenzado.
INCLUIDO EN UN
PLAN DE VALIDACION: Se entenderá así a todo proceso o técnica, cuya validación
se encuentre incluida en el Programa del Plan de Validación.
• CLAUSURA:
ADMINISTRACION E
INFORMACION GENERAL
Clausura Total:
Ø Ausencia de
autorización del funcionamiento del establecimiento por la Autoridad Sanitaria competente.
Ø Falta de
solicitud (inicio de trámite ante la Autoridad Sanitaria) de inscripción del Director Técnico y del Codirector Técnico, cuando lo
hubiere, ante la Autoridad Sanitaria competente.
GARANTIA DE
CALIDAD
Clausura Total
Ø Inexistencia en
la empresa de un Programa de Garantía de Calidad.
Ø Incumplimiento
no justificado de los programas de Garantía de Calidad de la empresa.
• CLAUSURA Y
RETIRO DEL MERCADO ADMINISTRACION E INFORMACION GENERAL
Ø Ausencia de
registro (certificado de autorización/inscripción) de productos comercializados
en el país.
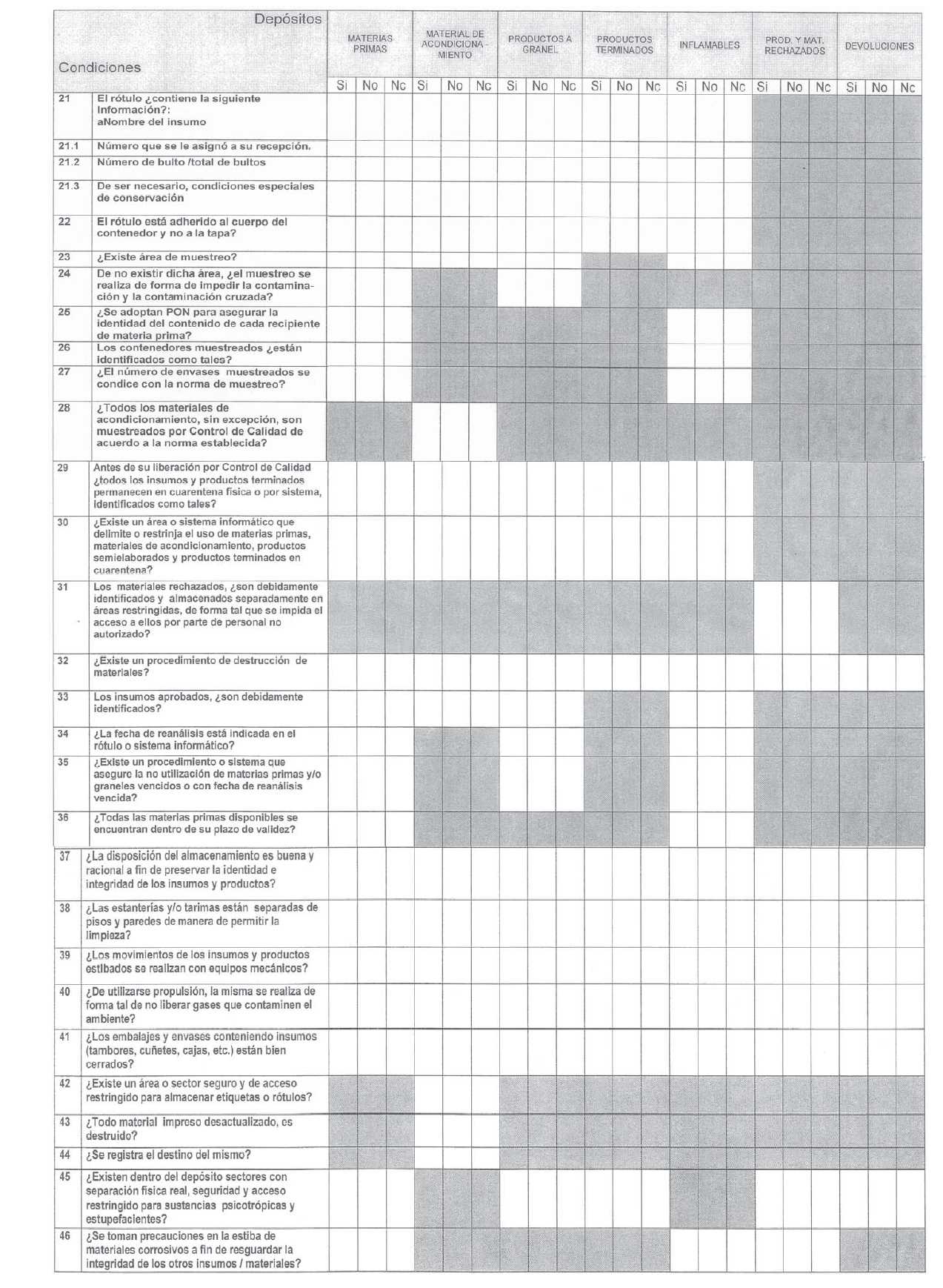
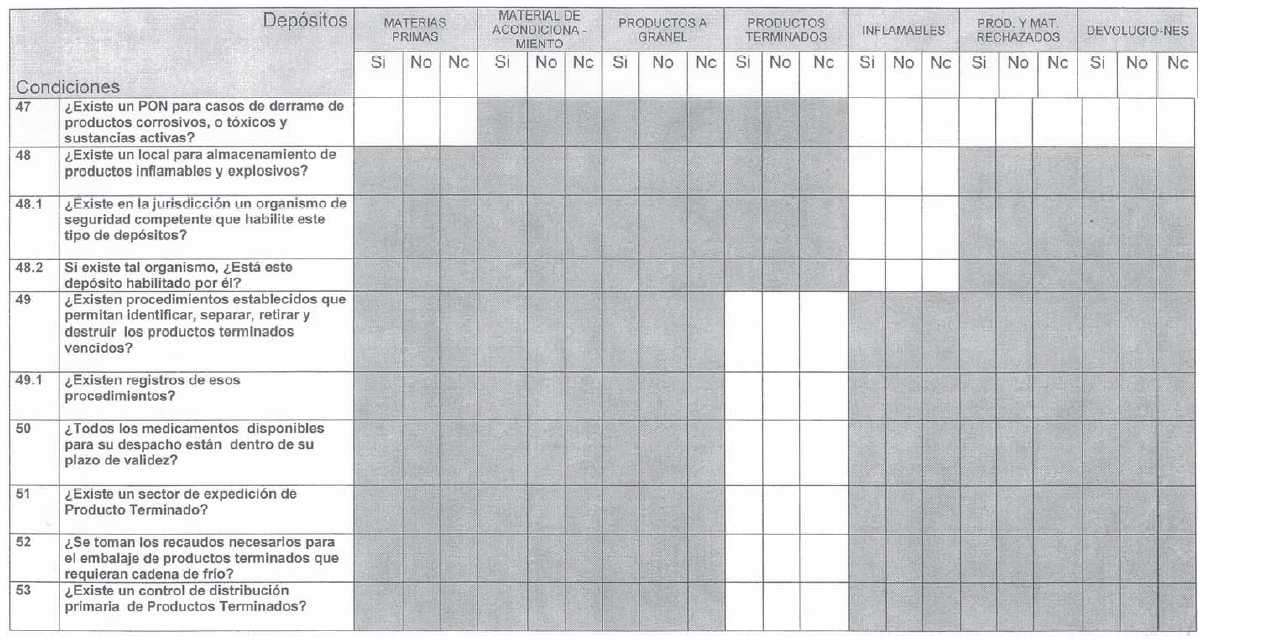
DEPOSITOS
Ø Productos
disponibles para su comercialización sin previa autorización de la Dirección Técnica o de profesional autorizado por la misma.
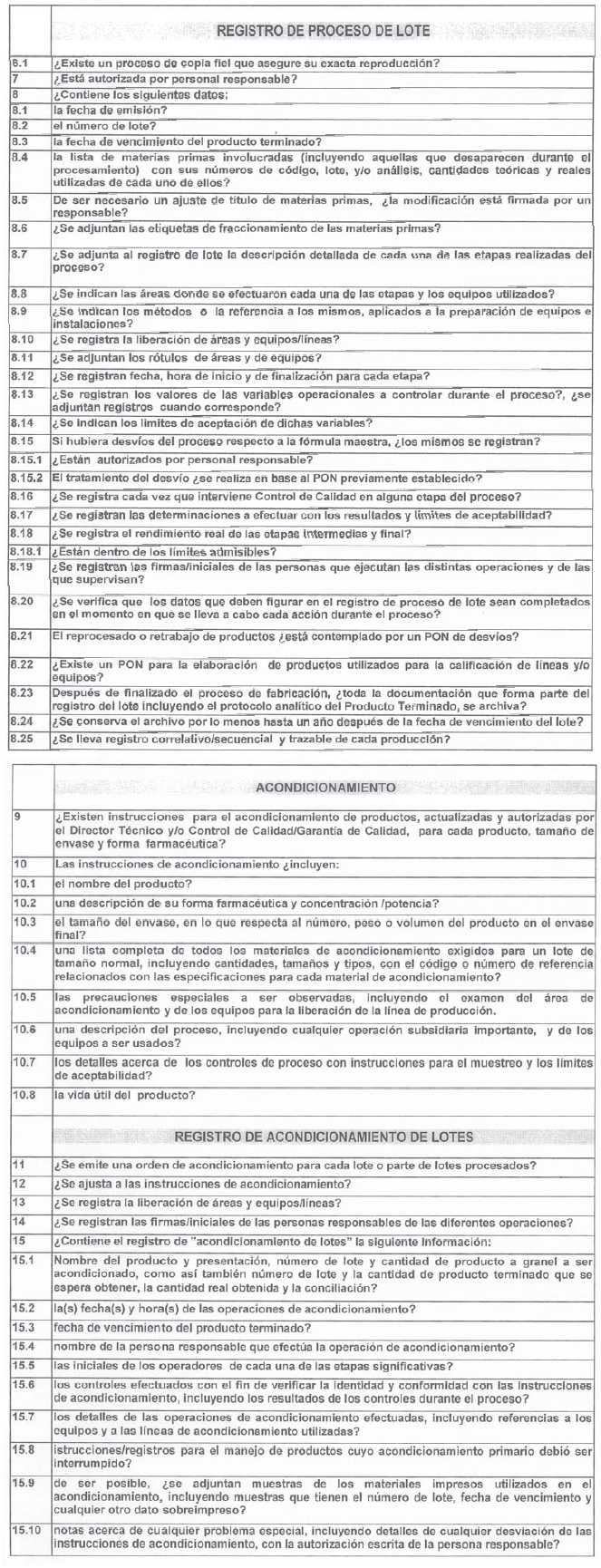
DOCUMENTACION DE LA PRODUCCION: Fórmula maestra:
Ø Evidencia de
adulteración de las órdenes de fabricación.
Ø Evidencia de
adulteración de registros.
DOCUMENTACION DE LA PRODUCCION: Registro de proceso de lote
Ø Ausencia de
registros de proceso de lotes (batch records)
PRODUCCION:
Equipamiento
Ø Evidencia de
contaminación de materias primas, materiales de acondicionamiento primario,
productos semielaborados y productos terminados aprobados con materiales
extraños, como grasa, aceites, herrumbre, partículas o elementos provenientes
de los equipos, agentes fumigantes y rodenticidas.
CONTROL DE CALIDAD
Ø Evidencia de
adulteración de datos y/o resultados analíticos.
Ø Falta de control
de calidad, según especificaciones, antes de la liberación de los productos
terminados para la venta.
Ø Falta de control
de calidad de materias primas y de materiales de envases y empaque
• INHIBICION
ADMINISTRACION E
INFORMACION GENERAL
Ø Ausencia del
Director Técnico, del Codirector Técnico y del profesional responsable según
organigrama estando la planta en actividad.
RECURSOS HUMANOS:
Ø Falta de
independencia entre Control de Calidad y Producción.
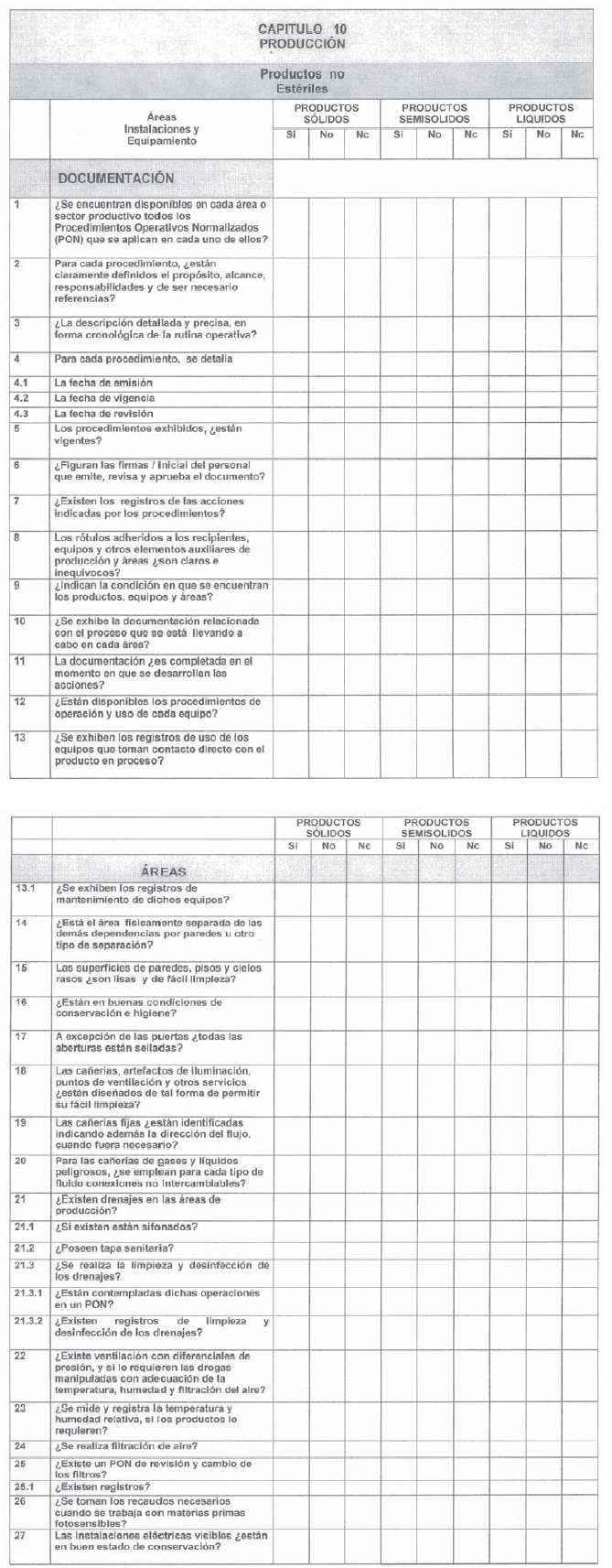
INSTALACIONES:
Condiciones Generales
Ø Evidencia de
acumulación de residuos / materiales extraños que indique falta de limpieza en
áreas de fabricación.
Ø Inexistencia de
sistema de filtración de aire para evitar la contaminación del ambiente con
materias primas y/o productos, que pueda generarse durante las actividades de
producción.
Ø Deficiente
funcionamiento del sistema de manejo de aire que puede generar una posible
contaminación cruzada.
Ø Producción de
productos diferentes, en forma simultánea en un mismo ambiente que pueda
conducir a mezclas o contaminación cruzada entre productos.
Ø Ausencia de
vestuarios generales de planta separados de las áreas de producción y control
y/o falta de provisión de la vestimenta de trabajo.
Ø Uso de las
instalaciones para fines no autorizados por la Autoridad Sanitaria competente.
SISTEMAS E
INSTALACIONES DE AGUA
Ø Calidad de agua
reiteradamente fuera de especificaciones de Farmacopeas debido a deficiente
mantenimiento u operación del sistema de tratamiento.
DEPOSITOS
Ø Ausencia de
responsable designado (perteneciente a la empresa o contratado) de higiene y
seguridad industrial.
Ø Area de muestreo
de materias primas no separada o precauciones insuficientes para prevenir la
contaminación y la contaminación cruzada durante el muestreo de materias
primas.
Ø Inadecuadas
prácticas de manejo de productos en cuarentena, rechazados o retirados del
mercado que permitan su comercialización.
Ø Ausencia de
registros de distribución primaria de productos terminados.
DOCUMENTACION DE LA PRODUCCION
Ø Falta de
registros que evidencien el uso adecuado de material de acondicionamiento
codificado y no codificado (incluyendo el almacenamiento, distribución,
impresión y descarte)
Ø Falta de
identificación propia de materiales en proceso y de áreas de producción
generando una alta probabilidad de mezclas.
Ø Ausencia de
documentación que certifique la inscripción / habilitación por parte de la Autoridad Sanitaria competente de los laboratorios terceristas contratados y los contratos
correspondientes.
CENTRAL DE PESADAS
Ø Falta de área
físicamente separada para central de pesadas.
Ø Falta de
sistemas para la extracción localizada de polvos y filtración de aire.
PRODUCCION:
Productos Segregados
Ø Falta de
independencia y autonomía de áreas de producción de productos segregados,
respecto de otras áreas de producción (ya sea de productos segregados o no
segregados).
Ø Uso de
vestimenta no exclusiva para la elaboración de estos productos.
PRODUCCION:
Operaciones
Ø Falta de plan de
validación de procesos de producción.
CONTROL DE CALIDAD
Ø No se asegura la
identidad del contenido de cada recipiente de materia prima antes de su uso.
Ø Falta la persona
responsable asignada.
Ø El Departamento
de Control de Calidad no es una unidad distinta e independiente de producción,
careciendo del real poder de decisión.
Ø Para
laboratorios de ensayo propios, falta de un programa de calificación,
calibración y mantenimiento de equipos y/o falta de un programa de preparación
y sistema de mantenimiento para estándares y para soluciones, y/o falta de
mantenimiento de registros del cumplimiento de estos programas.
Ø Los métodos de
control de principios activos no codificados no están incluidos en el plan de
validación.
• INHIBICION Y
RETIRO DEL MERCADO
CONTROL DE CALIDAD
Ø Falta de un
programa y/o procedimiento de reanálisis de materias primas.
Ø No se realiza
control de calidad en laboratorio propio, en el país, de productos importados.
• INHIBICION DE
LINEA / AREA / LOTE
INSTALACIONES:
Condiciones Generales
Ø Servicios y
sistemas que puedan afectar la calidad del/los producto/s no incluidos en un
Plan de Calificaciones.
Ø Ausencia de
mantenimiento y de verificación periódica del sistema de manejo de aire, como
ser por ejemplo reemplazo de filtros y monitoreo de presiones diferenciales.
Ø La limpieza de
equipos y áreas de producción no estén incluidas en el plan de validaciones.
Ø Falta de
verificación de limpieza de áreas y equipos, cuando no ha sido validada.
Ø Falta de
limpieza de áreas entre el procesamiento de distintos productos.
Ø Falta de
limpieza entre el procesamiento de distintos lotes del mismo producto,
elaborados en días sucesivos, no avalado el lapso mayor por una validación de
limpieza.
Ø Instalaciones y
servicios con evidente falta de limpieza, ubicadas directamente encima de
productos expuestos o de los equipos de manufactura.
Ø Pisos, paredes y
cielos rasos, en áreas de manufactura, con evidencia de falta de limpieza.
Ø Existencia de
drenajes en las áreas de elaboración aséptica y llenado aséptico.
Ø Temperatura,
humedad y/o condiciones de iluminación no controladas o monitoreadas cuando se
requiere y/o fuera de especificaciones.
SISTEMAS E
INSTALACIONES DE AGUA: Agua Purificada
Ø No se hacen
ensayos microbiológicos los días de uso o con una frecuencia establecida
debidamente validada en el caso de sistemas de producción de agua purificada
continuos.
Ø No se hacen ensayos
microbiológicos los días de uso en el caso de sistemas de producción de agua
purificada no continuos.
Ø No se hacen
ensayos físico-químicos codificados en farmacopeas FE, USP, en la FA actualizada o según métodos alternativos validados para la liberación de cada lote, en el
caso de sistemas de producción de agua purificada no continuos.
Ø No se hacen
análisis físico-químicos diarios o con una frecuencia establecida según las
metodologías establecidas por las ediciones vigentes de la FE o USP, o actualizada de la FA, o según métodos alternativos validados, en el caso de
sistemas de producción de agua purificada continuos.
Ø Distribución de
agua purificada a través de cañerías de material no sanitario
SISTEMAS E
INSTALACIONES DE AGUA: Agua para Inyectables
Ø Falta de
mantenimiento de los sistemas de obtención de agua para inyectables.
Ø Sistemas de
obtención de agua para inyectables no acorde a lo codificado en las
metodologías establecidas por las ediciones vigentes de la FE o USP, o actualizada de la FA.
Ø Utilización del
agua para inyectables conservada más allá de la jornada de trabajo sin haberse
mantenido a más de 70º C o a menos de 4º C y con recirculación constante por un
anillo hasta los puntos de uso.
Ø Ausencia de plan
de validación del almacenamiento a esa temperatura (más de 70º C o a menos de
4º C).
Ø No se hacen
ensayos microbiológicos y de endotoxinas en los días de uso o con una
frecuencia establecida debidamente validada en el caso de sistemas de
producción de agua para inyectables continuos.
Ø No se hacen
ensayos microbiológicos y de endotoxinas de cada lote en el caso de sistemas de
producción no continuos de agua para inyectables.
Ø No se hacen
ensayos físico-químicos codificados en las ediciones vigentes de farmacopeas
(FE, USP, o FA actualizada) o según métodos alternativos validados para la
liberación de cada lote, en el caso de sistemas de producción no continuos de
agua para inyectables.
Ø No se hacen
análisis físico-químicos los días de uso según las metodologías codificadas en
las ediciones vigentes de la FE o USP, o actualizada de la FA, o según métodos alternativos validados, en el caso de sistemas de producción de agua para
inyectables continuos.
Ø Revalidación
inadecuada de los sistemas de obtención de agua para inyectables luego de
efectuar cambios que puedan afectar la calidad de la misma.
Ø No se utiliza
agua para inyectables para el enjuague final de los equipos o componentes
utilizados en la fabricación de productos parenterales.
CENTRAL DE PESADAS
Ø Falta de
adecuación de la temperatura, humedad, iluminación especial si lo requieren las
drogas manipuladas.
DOCUMENTACION DE LA PRODUCCION: Registro de Proceso de Lote
Ø Falta de PON que
establezca medidas para garantizar la trazabilidad de cada lote, en caso de
fabricarse simultáneamente diferentes lotes del mismo producto en una misma
área.
Ø Falta de
liberación de líneas de producción entre distintas elaboraciones o liberaciones
no documentadas.
Ø Falta de control
de los instrumentos críticos de medición o falta de documentación de esos
controles.
Ø Reprocesamiento
o retrabajo no contemplados en PON de desvíos.
PRODUCCION:
Equipamiento
Ø El equipamiento
útilizado en operaciones de fabricación complejas o de productos críticos, no
está incluido en el plan de calificaciones.
Ø Existencia de
máquinas comprimidoras instaladas en un mismo ambiente.
Ø Los materiales
empleados en la construcción de los equipos no son compatibles con los
principios activos que entran en contacto con ellos.
Ø Falta de
limpieza de elementos de manipulación de drogas.
Ø El equipamiento
para producción es de características no sanitarias (material poroso, de
difícil limpieza, liberador de partículas, etc.).
PRODUCCION:
Operaciones
Ø La transferencia
de graneles/semielaborados entre etapas de producción no se realiza de forma de
evitar la contaminación de los mismos.
Ø Los equipos como
horno de despirogenado, autoclave, estufas de secado, etc. contienen más de un
producto al mismo tiempo con posibilidad de contaminación cruzada o mezcla.
Ø Realización de
actividades ajenas a la fabricación en áreas de fabricación.
Ø Realización de
actividades de fabricación en áreas ajenas a ese fin.
Ø Existencia de
recipientes abiertos, fuera de la áreas de producción, conteniendo producto
semielaborado.
Ø Las suspensiones
y/o emulsiones no se mantienen homogéneas durante todo el proceso de
fraccionamiento.
PRODUCCION:
Productos no Estériles
Ø Falta de
monitoreo microbiológico y ambiental en áreas donde se elaboran productos no
estériles cuando el producto lo requiere.
PRODUCCION:
Productos Estériles
Ø Operaciones de
llenado aséptico realizadas luego de pruebas de llenado aséptico con medio de
cultivo, en las condiciones normales de trabajo, con resultados no
satisfactorios.
Ø Ciclos de
esterilización basados en probabilidad de supervivencia microbiana no validada.
Ø Sistemas de
obtención de agua para inyectables no validados y/o con reiterada evidencia de
recuentos microbiológicos y/o valores de endotoxinas fuera de especificaciones
sin registro de acciones correctivas tomadas.
Ø Falta de pruebas
de llenado aséptico con medio de cultivo, en las condiciones normales de
trabajo y siempre que sea técnicamente factible, para demostrar la validación
de las operaciones de llenado aséptico.
Ø Ambientes no
clasificados y no controlados y falta de monitoreo de microorganismos viables
durante la elaboración de productos de llenado aséptico.
Ø Programa de
sanitización / desinfección faltante o incompleto o existencia de desvíos
microbiológicos sostenidos.
Ø Falta de
esterilización de los envases para productos oftálmicos.
Ø Interpretación
errónea de los resultados del llenado con medios de cultivo o no realización de
estos ensayos.
En las áreas de
ambiente controlado la vestimenta no es la exigida para cada grado de aire.
Ø Número
insuficiente de unidades llenadas durante el llenado con medios de cultivo.
Ø El llenado con
medios de cultivo no simula operaciones reales.
Ø En las pruebas
de llenado aséptico, no está demostrada la capacidad del medio para favorecer
el desarrollo de un amplio espectro de microorganismos.
Ø No se realiza el
ensayo de endotoxinas en el agua para inyectables utilizada en el enjuague
final de envases y componentes utilizados para medicamentos parenterales,
cuando dichos envases y componentes no son luego despirogenados.
Ø Falta de
registradores continuos de tiempo y temperatura en los equipos de
despirogenado.
Ø Falta de
registradores continuos de tiempo y temperatura en los equipos de autoclavado.
Ø Falta de área
separada para el lavado, esterilización y despirogenado de ampollas y/o frascos
ampolla vacíos y tapones.
Ø En las maquinas
lavadoras de frascos y ampollas vacíos, no se utiliza agua para inyectables, al
menos para el último enjuague.
Ø Falta de
validación de los ciclos de despirogenado.
Ø Uso de
vestimenta no exclusiva en las áreas de ambiente controlado.
Ø El ingreso del
personal al área de llenado no se realiza a través de acceso directo desde el
vestuario para áreas limpias.
Ø En los ciclos de
esterilización se usa vapor que contiene aditivos en un nivel tal que puede ser
causa de contaminación del producto o de los equipos.
Ø El agua
utilizada en los autoclaves para el enfriamiento mediante lluvia de las
Soluciones Parenterales de Gran Volumen contiene aditivos en un nivel tal que
puede ser causa de contaminación del producto o de los equipos.
Ø Falta de
monitoreo microbiológico y ambiental en áreas donde se elaboran productos
estériles.
Ø Falta de
monitoreo microbiológico y ambiental en cada operación aséptica.
Ø Volumen
insuficiente de las muestras de aire tomadas para la clasificación de áreas
metodología de muestreo inadecuada.
Ø Instalaciones o
equipos no mantenidos para minimizar contaminación / generación de partículas.
Ø Las áreas de
ambiente controlado no cumplen con la clasificación exigida.
Ø Las áreas de
llenado de productos parenterales no cumplen con la clasificación de grado A
con entorno al menos grado C, si no se utilizan equipos de
soplado-llenado-sellado ni aisladores.
Ø Falta de
inyección de aire filtrado en los vestuarios de las áreas de ambiente
controlado.
Ø El vapor
utilizado en la esterilización no es monitoreado para asegurar que no contiene
aditivos en cantidad tal que sea fuente de contaminación del producto o de los
equipos.
Ø Falta de PON que
establezca el número máximo de personal que puede estar presente en Areas
Limpias y asépticas mientras están en operación.
Ø La cantidad de
personal en las áreas limpias y asépticas, mientras están en operación, supera
al especificado en el PON.
Ø Incorporación de
gases, sin previo paso por filtro esterilizante, a soluciones esterilizadas.
Ø Inspección de
partículas y defectos críticos no realizada sobre el 100 % de las unidades.
CONTROL DE CALIDAD
Ø Para
laboratorios de ensayo contratados, falta de un programa de calificación,
calibración y mantenimiento de equipos y/o falta de un programa de preparación
y sistema de mantenimiento para estándares y para soluciones, y/o falta de
mantenimiento de registros del cumplimiento de estos programas, en todos los
casos aquellos que involucren a los ensayos contratados.
GARANTIA DE
CALIDAD
Ø Revalidación de
cualquier proceso que ha sufrido una modificación crítica que pueda influir en
la reproducibilidad del mismo y/o en la calidad del producto final, no incluida
en el plan de validaciones.
GARANTIA DE
CALIDAD: Estabilidad
Ø Inexistencia de
un programa de seguimiento de estabilidad de los productos comercializados.
• INHIBICION DE
LINEA / AREA / LOTE Y RETIRO DEL MERCADO
DOCUMENTACION DE LA PRODUCCION: Fórmula Maestra
Ø Falta de Fórmula
Maestra.
Ø Fórmula maestra
no actualizada y/o no autorizada por el Director Técnico y Garantía de Calidad
para cada producto y tamaño de lote a fabricarse.
Ø No emisión de
órdenes de producción o existencia de órdenes de producción no coincidentes con
las formulas maestras según Disposición 2819/04 y sus actualizaciones.
DOCUMENTACION DE LA PRODUCCION: Registro de Proceso de Lote
Ø El batch record
muestra significativos errores de cálculo.
Ø Cambios en las
operaciones críticas no incluidos en el plan de validación o información no
disponible.
Ø Cambios en las
operaciones críticas (comparados con los documentos de producción maestra) no
aprobados por Garantía de Calidad y Dirección Técnica, o no documentados.
Ø Desviaciones no
documentadas durante las elaboraciones.
Ø Desviaciones no
aprobadas por Garantía de Calidad y/o Dirección Técnica.
Ø Discrepancias en
los rendimientos y conciliaciones no investigadas.
DEPOSITOS:
Ø Falta de
recaudos para el depósito y/o embalaje de productos terminados que requieran
cadena de frío.
Ø Distintos lotes
de materia prima ingresados al mismo tiempo y no muestreados y analizados
independientemente.
Ø Productos
terminados liberados para su uso y elaborados con materias primas vencidas.
Ø Productos
terminados liberados para su uso y elaborados con materias primas con fecha de
reanálisis vencida.
INSTALACIONES:
Condiciones Generales
Ø Mal
funcionamiento del sistema de manejo de aire con evidencia de contaminación
cruzada.
Ø Areas y/o
equipos de manufactura con evidencia de contaminación (polvos, hongos, restos
de producciones anteriores, etc.).
Ø Daños (agujeros,
grietas o descascaramiento de pintura) en paredes, piso y/o cielos rasos de
áreas de manufactura en los cuales el producto esta expuesto.
SISTEMAS E
INSTALACIONES DE AGUA: Agua Purificada
Ø No uso de agua
purificada como materia prima para la elaboración de productos no parenterales.
Ø Calidad no
aceptable del agua usada en la preparación.
PRODUCCION:
Productos Estériles
Ø Elaboración de
productos parenterales con agua de calidad no para inyectables.
Ø Ausencia de
ensayo muestral de hermeticidad de ampollas.
Ø Cada carga de
estilización del producto envasado no es considerada como un lote separado para
el ensayo de esterilidad.
CONTROL DE
CALIDAD:
Ø Producto
terminado liberado para su uso, con control de calidad incompleto o con
resultados de análisis que no se ajustan a las especificaciones establecidas.
Ø Uso de
materiales de partida sin autorización de control de calidad.
• RETIRO DEL
MERCADO
PRODUCCION:
Operaciones
Ø La información
impresa o codificada en los envases primarios del lote de producción no es
legible y/o se destiñe y/o se borra con facilidad por manipuleo.
Ø Falta de número
de lote y fecha de vencimiento en los envases primarios de los productos
terminados.
CONTROL DE CALIDAD
Ø Número de
muestras para ensayo de esterilidad inferior al definido en la edición vigente
de la Farmacopea Argentina o de las farmacopeas reconocidas internacionalmente
(FE, USP).
Ø Falta de control
de calidad individual de lotes y/o sublotes de producción.
Ø Productos que
caen fuera de especificaciones dentro de su período de vida útil.
GARANTIA DE
CALIDAD
Ø Desviaciones no
investigadas ni documentadas de conformidad con procedimientos escritos.
GARANTIA DE
CALIDAD: Estabilidad
Ø No son tomadas
acciones cuando los productos ya liberados para su comercialización caen fuera
de especificaciones dentro de su período de vida útil.
Ø No existen
programas de seguimiento de estabilidad relacionados con cambios en la
manufactura (formulación y/o cambios críticos en los métodos de elaboración)
y/o en los materiales de acondicionamiento primarios.
• MEDIDA
CORRECTIVA INMEDIATA
RECURSOS HUMANOS
Ø Falta de
organigrama.
INSTALACIONES:
Condiciones Generales
Ø Drenajes de piso
sin rejillas o tapas.
Ø Salidas para
líquidos y gases sin identificar.
Ø No existencia de
normas o PON para limitar el ingreso de personas no autorizadas a áreas
restringidas.
Ø No cumplimiento
de las normas o PON para limitar el ingreso de personas no autorizadas a áreas
restringidas.
Ø Falta de orden
y/o higiene en baños y vestuarios.
SISTEMAS E
INSTALACIONES DE AGUA
Ø Falta de
controles del agua potable.
Ø Falta de PON de
mantenimiento y/o de procedimiento de manejo de sistema de agua.
DEPOSITOS
Ø Cuarentena
física o informatizada accesible a personal no autorizado.
Ø Area de
cuarentena física no bien demarcada y/o no respetada si no se dispone de un
sistema de cuarentena informatizada.
Ø Falta de
verificación, según PON, en el ingreso de materiales.
Ø Incompleta
identificación de materiales, graneles, materias primas, productos terminados.
Ø Presencia de
medicamentos vencidos junto a aquellos disponibles para la venta.
Ø Existencia de
material de envase/empaque obsoleto o fuera de validez no aislado e
identificado.
Ø Falta dentro del
depósito de sectores con separación física real, seguridad y acceso restringido
para sustancias psicotrópicas y estupefacientes.
DEVOLUCIONES
Ø No existe PON
para el manejo de productos devueltos.
DOCUMENTACION DE LA PRODUCCION:
Ø Falta rotulación
de las drogas pesadas o medidas que incluya nombre o código de la materia prima
y nombre o código del producto al que se destina.
Ø Tiempo de
retención de registros de proceso de lotes menor a un año después de la fecha
de vencimiento del producto.
Ø Registros de
programas de limpieza y sanitización incompletos.
Ø Correcciones en
la documentación, realizadas con corrector o goma de borrar y/o sin registro de
fecha y firma.
Ø Fórmulas
maestras verificadas por personal no autorizado.
Ø Cambios no
autorizados en los tamaños de lote.
Ø Desvíos durante
el acondicionamiento no investigados por personal calificado.
PRODUCCION:
Equipamiento
Ø Equipamiento
defectuoso o fuera de uso inadecuadamente rotulado o no retirado del área.
Ø Los equipos
limpios no están protegidos contra la contaminación.
Ø Falta de programa
de calibración de equipos de medida automáticos, mecánicos o electrónicos.
Ø Falta de
bitácora de uso de equipos (log books).
PRODUCCION:
Operaciones
Ø Existencia de
envases primarios sobrantes codificados con número de lote y/o fecha de
vencimiento no aislados ni identificados.
Ø Acceso a los
vestuarios específicos de áreas de producción/fraccionamiento sin la vestimenta
protectora de planta.
PRODUCCION:
Productos Estériles
Ø Falta de evidencia
documentada de destrucción de material descartado (ampollas, materiales de
envase y empaque, etc.).
Ø Productos
esterilizables por calor húmedo (autoclavables) que no han sido esterilizados
por este método.
CONTROL DE CALIDAD
Ø Almacenamiento
de muestras fuera de sus especificaciones de conservación.
Ø Utilización en
producción de lotes de materias primas sin autorización de Control de Calidad.
Ø Especificaciones
incompletas y/o no aprobadas por Garantía de Calidad y/o Dirección Técnica.
Ø Ausencia de
ensayos de materiales de acondicionamiento, sin que se haya calificado a los
proveedores.
Ø No se guardan
muestras de retención de productos terminados y/o de materias primas.
• MEDIDA
CORRECTIVA MEDIATA
ADMINISTRACION E
INFORMACION GENERAL
Ø Falta de
concordancia entre la estructura edilicia observada y el plano aprobado por la Autoridad Sanitaria competente.
INSTALACIONES:
Condiciones Generales
Ø La distancia
entre equipos y paredes es insuficiente para permitir la limpieza.
Ø La base de los
equipos inmóviles no están debidamente fijada a puntos de apoyo.
Ø Ausencia de
Programa de Sanitización escrito aunque las instalaciones se observan en
adecuadas condiciones de limpieza.
Ø Programa escrito
de sanitización incompleto aunque las instalaciones se observan en estado de
limpieza aceptable.
Ø Ausencia de
prevención contra la entrada y/o proliferación de roedores, insectos, aves u
otros animales.
Ø Si bien se
observan daños en superficies, los productos no se encuentran directamente
expuestos a las mismas (ej. pared de depósito de producto terminado con pintura
descascarada).
Ø Falta de
mantenimiento en baños y vestuarios.
SISTEMAS E
INSTALACIONES DE AGUA: Agua Purificada
Ø No uso de agua
purificada para el enjuague final de los equipos usados para la elaboración de
productos no parenterales.
Ø Falta de
mantenimiento de los sistemas de obtención de agua purificada, con calidad de
agua dentro de especificaciones.
RETIRO DE
PRODUCTOS DEL MERCADO
Ø Falta de PON que
establezca el sistema de retiro de productos del mercado o el procedimiento no
contempla la obligación de comunicación inmediata a la Autoridad Sanitaria competente.
DOCUMENTACION DE LA PRODUCCION
Ø Manipulación de
materias primas realizada por personas no autorizadas.
Ø Falta de
procedimiento escrito para las operaciones de acondicionamiento.
PRODUCCION:
Equipamiento
Ø Los tanques para
la elaboración de líquidos y semisólidos no tienen diseño / material sanitario.
PRODUCCION:
Productos Estériles
Ø Falta de PON
para el control microbiológico de áreas limpias.
Ø No es tenida en
cuenta la carga microbiana para la validación de los ciclos de esterilización.
Ø Operarios de
inspección de partículas y defectos no rotados durante toda la jornada, o sin
control oftalmológico periódico.
Ø Lapsos de
tiempos entre limpieza, esterilización, uso de componentes, contenedores y
equipos, no incluidos en el programa de validaciones.
Ø Lapsos de tiempo
entre el comienzo de la manufactura y la esterilización o filtración
esterilizante, no incluidos en el programa de validaciones si excede las ocho
horas.
CONTROL DE CALIDAD
Ø No existen PON
para el muestreo, inspección y ensayo de materiales.
GARANTIA DE
CALIDAD
Ø Ausencia de un
sistema de control de cambios.
Ø Validación
incompleta de metodologías de análisis no codificadas.
Ø Falta de
programa de autoinspecciones, o programa de autoinspecciones que no abarca
todas las secciones de GMP o falta de registros o con registros incompletos.
Ø No existe un
sistema para el manejo de quejas y reclamos.
Ø Programa de
ensayo reducido puesto en práctica sin calificación de proveedores.
Ø Calificación de
proveedores no documentada.
• MEDIDA
CORRECTIVA PROGRAMADA
RECURSOS HUMANOS
Ø Ausencia de
programa y registro de capacitación del personal en BPFyC.
INSTALACIONES:
Condiciones Generales.
Ø El personal
utiliza puertas de acceso directo del exterior hacia áreas de producción y/o
empaque.
DOCUMENTACION DE LA PRODUCCION
Ø Procedimientos
escritos incompletos para manejo de materiales y productos.
Ø Procedimientos incompletos
para las operaciones de empaque.
PRODUCCION:
Equipamiento
Ø Uso de equipos
menores no calificados para la manufactura de productos no críticos.
CONTROL DE CALIDAD
Ø Falta de ensayos
de identificación, realizados por el titular del producto, sobre los remanentes
de materia prima luego de su uso por terceros.
GARANTIA DE
CALIDAD: Estabilidad
Ø No se sigue el
programa de seguimiento de estabilidad de productos comercializados.
Ø No se controlan
todos los parámetros que pueden ser afectados por la estabilidad del producto.
GARANTIA DE
CALIDAD: Auditoría a Proveedores
Ø Falta de
programa de calificación de proveedores de materia prima inactiva y de
materiales de acondicionamiento.