Administración Nacional de Medicamentos, Alimentos y
Tecnología Médica
SALUD PUBLICA
Disposición 2819/2004
Apruébanse los lineamientos generales de Buenas Prácticas
de Fabricación para Elaboradores, Importadores/Exportadores de Medicamentos.
Bs. As., 18/5/2004
VISTO la Ley nº 16.643, sus Decretos Reglamentarios nros.
9.763/64, 150/92 y 177/93 y los Decretos nros. 1.490/92 y 341/92; y el
Expediente nº 1-47-1110-1281-04-0 del Registro de esta Administración Nacional;
y
CONSIDERANDO;
Que la fiscalización de los Establecimientos Productores,
Importadores y Distribuidores de Especialidades Medicinales, a través de
inspecciones técnicas, es un mecanismo idóneo que contribuye a garantizar la
calidad con que llegan al mercado los productos que elaboran, importan y
distribuyen esos establecimientos.
Que dicha fiscalización debe cubrir aspectos relativos a
condiciones de funcionamiento y sistemas de control de calidad utilizados por
los establecimientos alcanzados por la normativa referida precedentemente
Que las acciones de control son responsabilidad de esta
Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT),
quien debe contar con un modelo que asegure: a) el control de las industrias
con uniformidad de criterio y b) la neutralidad, simetría y reciprocidad en el
tratamiento y aplicación de las normas de regulación.
Que mediante la Disposición (ANMAT) nº 853/99 adoptaron las recomendaciones sobre Buenas Prácticas de Fabricación para la Industria Farmacéutica aprobadas por la Asamblea Mundial de la Salud en mayo de 1992.
Que como consecuencia de los avances tecnológicos resulta
necesario adoptar las Recomendaciones sobre Buenas Prácticas de Fabricación y
Control de 2003, aprobadas por la Asamblea Mundial de la Salud e informes de la PIC’S –Pharmaceutical Inspection Corporation Scheme-: PE 009-1 e ICH
–International Conference on Harmonisation - Guía de GMP – Q7A.
Que el Instituto Nacional de Medicamentos y la Dirección de Asuntos Jurídicos han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades conferidas por
los Decretos nros. 1490/92 y 197/02.
Por ello;
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1º — Apruébanse los lineamientos generales de Buenas
Prácticas de Fabricación para Elaboradores, Importadores / Exportadores de
Medicamentos y sus anexos, que a continuación se detallan y forman parte
integral de la presente Disposición:
Anexo I: Aplicación de la Metodología de Análisis de Peligros y Puntos Críticos de Control en la Producción de Medicamentos.
Anexo II: Calificación y Validación.
Anexo III: Liberación Paramétrica.
Anexo IV: Estándares para Ensayos Físico - Químicos.
Anexo V: Calificación de Proveedores de materiales de acondicionamiento
para la Industria Farmacéutica.
Anexo VI: Buenas Prácticas de Fabricación para
Ingredientes Farmacéuticos activos.
Anexo VII: Buenas Prácticas de Fabricación de
Preparaciones Radiofarmacéuticas.
Anexo VIII: Productos Fitoterápicos.
Anexo IX: Normas para la identificación por colores de
envases de las drogas para uso anestesiológico y de las soluciones
parenterales.
Anexo X: Buenas Prácticas de Fabricación de Productos
Medicinales derivados de la sangre o del plasma.
Anexo XI: Buenas Prácticas de Fabricación de Productos
Medicinales Biológicos para uso humano.
Art. 2º — Regístrese, comuníquese a quienes corresponda. Dése a la Dirección Nacional del Registro Oficial para su publicación; Cumplido, archívese PERMANENTE.
— Manuel R. Limeres.
Buenas Prácticas de Fabricación para Elaboradores,
Importadores/Exportadores de Medicamentos
ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y
TECNOLOGIA MEDICA
ANMAT
INSTITUTO NACIONAL DE MEDICAMENTOS
INAME
CONTENIDO
CONSIDERACIONES GENERALES
GLOSARIO
GESTION DE LA CALIDAD EN LA INDUSTRIA FARMACEUTICA: FILOSOFIA Y ELEMENTOS ESENCIALES
1. GARANTIA DE LA CALIDAD
2. BUENAS PRACTICAS DE FABRICACION PARA PRODUCTOS
FARMACEUTICOS (BPF)
3. SANITIZACION E HIGIENE
4. CALIFICACION Y VALIDACION
5. RECLAMOS
6. RETIRO DE PRODUCTOS
7. CONTRATO DE PRODUCCION Y ANALISIS
Generalidades
Contratante, Contratado y Contrato
8. AUTOINSPECCION Y AUDITORIAS DE CALIDAD
Elementos para la autoinspección
Equipo de autoinspección
Frecuencia de autoinspección y Reporte de autoinspección
Acciones de seguimiento
Auditorías de calidad
Auditorías de proveedores y aprobación
9. PERSONAL
Generalidades
Personal clave
10. ENTRENAMIENTO
11. HIGIENE PERSONAL
12. LOCALES
Generalidades
Areas auxiliares
Areas de depósito, pesada, producción y control de calidad
13. EQUIPAMIENTO
14. MATERIALES
Generalidades
Materias primas
Material de acondicionamiento
Productos intermedios, a granel y terminado
Materiales rechazados y recuperados
Productos retirados del mercado y devoluciones
Reactivos y medios de cultivo
Sustancias y materiales de referencia
Materiales de desecho
15. DOCUMENTACION
Generalidades
Documentos requeridos
16. BUENAS PRACTICAS DE PRODUCCION
Generalidades
Prevención de contaminación durante la producción
Operaciones de elaboración y acondicionamiento
17. BUENAS PRACTICAS DE CONTROL DE CALIDAD
Control de materias primas y productos intermedios, a
granel y terminados
Requisitos de los controles
Revisión del registro de lote
Estudios de estabilidad
18. PRODUCTOS FARMACEUTICOS ESTERILES
Introducción
Generalidades
Elaboración de productos estériles
Proceso
Esterilización
Personal
Locales
Equipos
Sanitización
Acabado de productos estériles
Control de calidad
ANEXO I
APLICACION DE LA METODOLOGIA DE ANALISIS DE PELIGROS Y PUNTOS CRITICOS DE CONTROL EN LA PRODUCCION DE MEDICAMENTOS
ANEXO II
CALIFICACION Y VALIDACION
ANEXO III
LIBERACION PARAMETRICA
ANEXO IV
DESARROLLO DE ESTANDARES PARA ENSAYOS FISICO-QUIMICOS
ANEXO V
CALIFICACION DE PROVEEDORES DE MATERIAL DE
ACONDICIONAMIENTO PARA LA INDUSTRIA FARMACEUTICA
ANEXO VI
BUENAS PRACTICAS DE FABRICACION PARA INGREDIENTES
FARMACEUTICOS ACTIVOS – (APIs)
ANEXO VII BUENAS PRACTICAS DE FABRICACION DE PREPARACIONES RADIOFARMACEUTICAS
ANEXO VIII BUENAS PRACTICAS DE FABRICACION DE MEDICAMENTOS
FITOTERAPICOS
ANEXO IX NORMAS PARA LA IDENTIFICACION POR COLORES DE ENVASES DE LAS DROGAS DE USO ANESTESIOLOGICO Y DE LAS
SOLUCIONES PARENTERALES
ANEXO X BUENAS PRACTICAS DE FABRICACION DE PRODUCTOS MEDICINALES
DERIVADOS DE LA SANGRE O DEL PLASMA HUMANO
ANEXO XI BUENAS PRACTICAS DE FABRICACION DE PRODUCTOS MEDICINALES
BIOLOGICOS PARA USO HUMANO
CONSIDERACIONES GENERALES
Los productos farmacéuticos autorizados para la comercialización,
deben ser fabricados sólo por las industrias autorizadas por la A.N.M.A.T, cuyas actividades son inspeccionadas regularmente por la Autoridad Sanitaria Nacional competente.
Las Buenas Prácticas de Fabricación (BPF) son aplicables a
las operaciones de fabricación de medicamentos en sus formas farmacéuticas
definitivas, incluyendo los procesos a gran escala en hospitales y la
preparación de suministros para el uso de ensayos clínicos.
Las Buenas Prácticas detalladas a continuación serán consideradas
lineamientos generales, y ellas son adoptadas para cumplir las necesidades
individuales. Las BPF como un todo, no cubren los aspectos de seguridad para el
personal comprometido en la fabricación, ni para la protección ambiental; éstos
están normalmente regulados por la Autoridad Nacional competente. Está también incorporado un nuevo concepto de análisis de
peligros relacionados a la producción y seguridad del personal (Anexo I). El
elaborador deberá garantizar la seguridad de los trabajadores y tomar las
medidas necesarias para prevenir la contaminación del medio ambiente.
GLOSARIO
Las definiciones dadas a continuación se aplican a los
términos utilizados en esta guía, éstas pueden tener significados diferentes en
otros contextos.
Acondicionamiento
Todas las operaciones, incluyendo el llenado y el
rotulado, que un producto a granel tiene que pasar para convertirse en producto
final. El llenado de un producto estéril bajo condiciones asépticas o un
producto que va a ser esterilizado en forma terminal, normalmente no se
considera como parte del acondicionamiento.
Area dedicada
Area que provee una separación completa y total de todos
los aspectos de una operación, incluyendo el movimiento del personal y
equipamientos, con procedimientos bien establecidos, controles y monitoreo.
Esto incluye barreras físicas como también sistemas de manejo de aire
separados, pero no necesariamente implica que deba encontrarse en un edificio
distinto y separado.
Area limpia
Area con control ambiental definido de partículas y
contaminación microbiana, construida y usada tal que se reduzca la
introducción, generación, y retención de contaminantes dentro del área.
Autorización de comercialización
Documento legal emitido por la Autoridad Sanitaria Nacional competente, que establece la composición y formulación detallada
del producto y las especificaciones de farmacopea u otras especificaciones
reconocidas de sus ingredientes y del producto final, e incluye detalles de
materiales de acondicionamiento, rótulos y vida útil.
Calibración
Conjunto de operaciones que establecen, bajo condiciones
específicas, la relación entre los valores indicados por un instrumento o
sistema de medición o los valores representados por una medida de un material,
y los correspondientes valores conocidos de un estándar de referencia, todos
registrados y controlados. Los límites de aceptación del resultado de medición
deben estar establecidos.
Calificación
Acción para evidenciar que las áreas, sistemas y equipos
trabajan correctamente y que finalmente conducen a los resultados esperados. El
sentido de la palabra "validación" a veces se extiende para
incorporar el concepto de calificación.
Conciliación
Comparación entre la cantidad teórica de producto o
materiales y la cantidad real producida o usada, permitiendo una variación
normal previamente asignada.
Contaminación
Introducción indeseada de impurezas de naturaleza química
o microbiológica, o de sustancias extrañas, dentro o sobre la materia prima o
productos intermedios durante la producción, muestreo, acondicionamiento,
almacenamiento o distribución.
Contaminación cruzada
Contaminación de una materia prima, producto intermedio, o
producto terminado con otra materia prima o producto durante la producción.
Control de Calidad
Ver sección 17.
Controles en proceso
Controles realizados durante la producción para monitorear
y, si es necesario, ajustar el proceso para asegurar que el producto cumple con
sus especificaciones. El control del ambiente y equipamiento pueden también ser
contemplados como parte del control en proceso.
Cuarentena
Estado de las materias primas o materiales de
acondicionamiento, productos intermedios, a granel o terminados aislados
físicamente o por otros medios efectivos, mientras se espera una decisión para su
liberación o rechazo.
Elaboración/Fabricación
Todas las operaciones de transformación de materiales y
productos, producción, control de calidad, liberación, almacenamiento,
transporte y distribución de productos farmacéuticos, y los controles relacionados.
Elaborador/Fabricante
Establecimiento que lleva a cabo operaciones tales como
producción, acondicionamiento, re-acondicionamiento, rotulado y re-rotulado de
los productos farmacéuticos.
Esclusa
Espacio cerrado con dos o más puertas, que se interpone
entre dos o más ambientes, por ejemplo, de diferentes áreas limpias
clasificadas, con el propósito de controlar el flujo de aire entre estos
ambientes, cuando se necesita ingresar en ellas. Una esclusa está diseñada
tanto para el uso de personas como de insumos y/o equipamiento.
Especificación
Lista de requerimientos detallados con los cuales los
productos o materiales utilizados u obtenidos durante la elaboración deben
cumplir. Estos sirven como una base para la evaluación de calidad.
Fórmula maestra
Documento o conjunto de documentos que especifican las
materias primas con sus cantidades y los materiales de acondicionamiento, junto
con una descripción de los procedimientos y precauciones requeridos para
producir una cantidad específica de un producto terminado, tanto como también
las instrucciones de elaboración, incluyendo los controles en proceso.
Garantía de la calidad
Ver sección 1.
Ingrediente farmacéutico activo (API)
Cualquier sustancia o mezcla de sustancias que serán
utilizadas en la fabricación de una forma farmacéutica, y que una vez
utilizada, se transforma en un ingrediente activo de dicha forma farmacéutica.
Tales sustancias son utilizadas para proporcionar actividad farmacológica u
otro efecto directo en el diagnóstico, cura, alivio, tratamiento o prevención
de una enfermedad, o que afecte la estructura y función del cuerpo.
Lote
Cantidad definida de materia prima, material de
acondicionamiento o producto, elaborado en un proceso o serie de procesos de
forma tal que sea homogénea.
"A fines de control del producto terminado, un lote
de un producto farmacéutico comprende todas las unidades de una forma
farmacéutica producidas a partir de la misma masa inicial de materiales, y que
ha sufrido una única serie de operaciones de fabricación o una sola operación
de esterilización o, en caso de proceso de producción continua, todas las
unidades fabricadas en un período de tiempo determinado".
Nota:
Con el fin de realizar ciertas fases de la elaboración,
puede ser necesario dividir un lote en diversos sub-lotes, que se unen después
para constituir un lote final homogéneo. En caso de elaboración continua, el
lote debe corresponder a una fracción definida de la producción, caracterizada
por su homogeneidad prevista.
Materia prima
Toda sustancia de calidad definida utilizada en la
producción de un producto farmacéutico, pero excluyendo materiales de
acondicionamiento.
Material de acondicionamiento
Todo material, incluyendo material impreso, empleado en el
acondicionamiento de un producto farmacéutico, pero excluyendo cualquier otro
envase exterior usado en el transporte. Los materiales de acondicionamiento
pueden ser primarios o secundarios de acuerdo a si están o no destinados a
estar en contacto directo con el producto.
Número de Lote
Combinación distintiva de números y/o letras que
identifica inequívocamente un lote tanto en los rótulos, su registro de lote y
certificados de análisis correspondientes, etc.
Operación crítica
Operación en el proceso de fabricación que puede causar variación
en la calidad del producto farmacéutico.
Parenterales de gran volumen
Solución o emulsión inyectable en base acuosa, estéril y
apirogénica, destinada a la administración intravenosa, acondicionada en un recipiente
para un único uso, que contiene un volumen igual o mayor a 100 ml, esterilizada
terminalmente. Se consideran incluidas en esta definición infusiones
intravenosas, soluciones para irrigación, soluciones para diálisis peritoneal y
soluciones para hemofiltración. No se incluye ningún producto de origen
biológico.
Persona autorizada
Persona reconocida por la Autoridad Sanitaria Nacional, como Director Técnico y/o Co-Director Técnico del laboratorio
titular del registro, que tiene la responsabilidad de asegurar que cada lote
del producto terminado ha sido fabricado, analizado y aprobado para su
liberación.
Procedimiento operativo normatizado (PON)
Procedimiento escrito autorizado que contiene
instrucciones para llevar a cabo operaciones no necesariamente específicas para
un dado producto o material (operación de equipos, mantenimiento y limpieza,
validación, limpieza de áreas y control ambiental, muestreo e inspección).
Ciertos PON pueden ser usados para complementar las especificaciones maestras
del producto y la documentación de producción del lote.
Producción
Todas las operaciones involucradas en la preparación de un
producto farmacéutico desde la recepción de materiales, a través del procesado
y acondicionamiento hasta la obtención del producto terminado.
Producto a granel
Todo producto que ha completado todas las etapas del
proceso, pero sin incluir el acondicionamiento final.
Producto intermedio
Producto procesado parcialmente que debe atravesar aún más
etapas de fabricación, previo a convertirse en producto a granel.
Producto farmacéutico (medicinal)
Todo material o producto destinado al uso humano
presentado en su forma farmacéutica definitiva o como materia prima para el uso
en tal forma farmacéutica, que está sujeto al control de la legislación
farmacéutica en cuanto a la elaboración, exportación y/o importación.
Producto terminado
Forma farmacéutica final que paso por todos los estadios
de fabricación incluyendo el acondicionamiento en el envase final.
Registro de lote
Todos los documentos asociados con la fabricación de un
lote tanto del producto a granel como del producto terminado. Estos proveen la
historia de cada lote del producto y de todas las circunstancias pertinentes a
la calidad del producto final.
Registro Maestro
Documento o conjunto de documentos que sirven de base para
la documentación del lote.
Reprocesado
El retrabajo de todo o parte de un lote de producto de
calidad inaceptable proveniente de una etapa definida de producción, con el fin
de que su calidad pueda ser aceptada.
Validación
Acción documentada, en concordancia con los principios de
las Buenas Prácticas de Fabricación, que demuestra que los procedimientos,
procesos, equipamientos, materiales, actividades o sistemas conducen realmente
a los resultados previstos. (Ver Calificación).
GESTION DE LA CALIDAD EN LA INDUSTRIA FARMACEUTICA: FILOSOFIA Y ELEMENTOS ESENCIALES
En la industria de medicamentos la gestión de calidad,
está comúnmente definida como el aspecto de la función de la gerencia que
determina e implementa la política de calidad, por ejemplo la intención global
y dirección de una organización en referencia a la calidad, formalmente
expresada y autorizada por la gerencia superior.
Los elementos básicos de la gestión de calidad son:
• Una infraestructura apropiada o sistema de calidad,
abarcando la estructura organizativa, procedimientos, procesos y recursos.
• Las acciones sistemáticas necesarias para asegurar la
confianza adecuada de que un producto o servicio satisfacerá los requerimientos
de calidad. La totalidad de éstas acciones son denominadas garantía de la
calidad.
Dentro de una organización, la garantía de la calidad
sirve como una herramienta gerencial. En situaciones contractuales, la garantía
de la calidad también sirve para generar confianza en el proveedor.
El concepto de Garantía de la Calidad, Buenas Prácticas de Fabricación y Control de Calidad son aspectos inter-relacionados
de la gestión de calidad. Ellos están descriptos aquí para enfatizar su
relación y su importancia fundamental en la producción y control de productos
farmacéuticos.
1. GARANTIA DE LA CALIDAD
1.1 Principio. "Garantía de la calidad" es un
concepto amplio que cubre todos los aspectos que individual o colectivamente
influyen en la calidad de un producto. Es la totalidad de las gestiones con el
objeto de asegurar que los productos farmacéuticos son de la calidad requerida
para su uso. Garantía de la calidad incorpora Buenas Prácticas de Fabricación y
otros factores incluyendo aquellos que están fuera del alcance de este Anexo,
tales como diseño y desarrollo de productos.
1.2 El sistema de garantía de calidad apropiado para el
elaborador de productos farmacéuticos debe asegurar que:
(a) los productos farmacéuticos son diseñados y desarrollados
en forma que condicen con los requerimientos de Buenas Prácticas de Fabricación
(BPF), Buenas Prácticas de Laboratorio (GLP) y Buenas Prácticas Clínicas (GCP),
(b) las operaciones de producción y control son claramente
especificadas en forma escrita y se adoptan los requerimientos de BPF,
(c) las responsabilidades gerenciales están claramente
especificadas en las descripciones de trabajo,
(d) las gestiones para la elaboración, provisión y uso de
las materias primas y materiales de empaque sean correctas,
(e) se llevan a cabo todos los controles necesarios en
materias primas, productos intermedio y a granel, controles en proceso,
calibraciones y validaciones,
(f) el producto final es correctamente elaborado y
controlado de acuerdo a procedimientos definidos,
(g) los productos farmacéuticos no son vendidos o
provistos antes que las personas autorizadas hayan certificado que cada lote de
producción ha sido producido y controlado de acuerdo con los requerimiento de
la autorización de comercialización y otras regulaciones relevantes a la
producción, control y liberación de productos farmacéuticos,
(h) las gestiones satisfactorias existen para asegurar
tanto como sea posible, que los productos farmacéuticos son almacenados por el
elaborador, distribuidos y subsecuentemente manejados tal que la calidad se
mantenga durante su vida útil,
(i) hay un procedimiento de autoinspección y/o auditoría
de calidad que regularmente evalúa la efectividad y aplicabilidad del sistema
de garantía de la calidad,
(j) los desvíos son reportados, investigados y
registrados,
(k) hay un sistema para aprobar cambios que pueden tener
un impacto sobre la calidad del producto,
(l) se deben llevar a cabo evaluaciones regulares de la
calidad de productos farmacéuticos con el objeto de verificar la consistencia
de los procesos y asegurar su mejora continua
1.3 El elaborador debe asumir la responsabilidad por la
calidad de los productos farmacéuticos, asegurando que los mismos sean aptos
para el uso previsto cumpliendo con los requerimientos de la autorización de
comercialización y que no pondrá en riesgo a los pacientes debido a seguridad,
calidad o eficacia inadecuadas. Las principales autoridades administrativas son
responsables del cumplimiento de este objetivo de calidad y requiere de la
participación activa y el compromiso de todos los departamento y a todos los
niveles dentro de la compañía, de los proveedores y distribuidores. Para que
sea posible alcanzar el mencionado objetivo de calidad se debe contar con un
sistema de Garantía de la Calidad de amplio alcance y correctamente aplicado,
que incorpore las Buenas Prácticas de Fabricación y Control de Calidad. Es
preciso que sea plenamente documentado y que su eficacia sea controlada. Todas
las partes del sistema de Garantía de la Calidad deben ser atendidas por personal competente y es necesario que se disponga de áreas, equipos e instalaciones
adecuadas.
2. BUENAS PRACTICAS DE FABRICACION PARA PRODUCTOS
FARMACEUTICOS (BPF)
2.1 Dentro del concepto de Garantía de Calidad, las Buenas
Prácticas de Fabricación constituyen el factor que asegura que los productos se
fabriquen en forma uniforme y controlada, de acuerdo con las normas de calidad
adecuadas al uso que se pretende dar a los productos, y conforme a las
condiciones exigidas para su comercialización. Las reglamentaciones que rigen
las BPF, tienen por objeto principal disminuir los riesgos inherentes a toda
producción farmacéutica. Dichos riesgos son esencialmente de dos tipos:
Contaminación (en particular de contaminantes inesperados) y mezclas
(confusión), causada, por ejemplo, por rótulos falsos colocados en envases. El
texto de las BPF exige:
(a) que todos los procesos de fabricación se definan
claramente, se revisen sistemáticamente a la luz de la experiencia, y se compruebe
que son el medio de fabricar productos farmacéuticos que tengan la calidad
adecuada para cumplir con las especificaciones;
(b) que se lleven a cabo calificaciones y validaciones;
(c) todos los recursos necesarios son provistos,
incluyendo:
I. Personal entrenado y apropiadamente calificado,
II. Instalaciones y espacios adecuados,
III. Servicios y equipamientos apropiados,
IV. Rótulos, envases y materiales apropiados,
V. Instrucciones y procedimientos aprobados,
VI. Transporte y depósito apropiados,
VII. Personal, laboratorios y equipamiento adecuado para
controles en proceso;
(d) que las instrucciones y procedimientos se redacten en
un lenguaje claro e inequívoco, que sean específicamente aplicables a los
medios de producción disponibles;
(e) que los operadores estén entrenados para efectuar
correctamente los procedimientos;
(f) que se mantengan registros (en forma manual o por
medio de aparatos de registro) durante la fabricación, para demostrar que todas
las operaciones exigidas por los procedimientos e instrucciones definidos han
sido en realidad efectuados y que la cantidad y calidad del producto son las
previstas; cualquier desviación significativa debe registrarse e investigarse
exhaustivamente;
(g) que los registros referentes a la fabricación y
distribución, los cuales permiten conocer la historia completa de un lote, se
mantengan de tal forma que sean completos y accesibles;
(h) que el almacenamiento y distribución de los productos
sean adecuados para reducir al mínimo cualquier riesgo de disminución de la
calidad;
(i) que se establezca un sistema que haga posible el
retiro de cualquier producto, sea en la etapa de distribución o de venta;
(j) que se estudie todo reclamo contra un producto ya
comercializado, como también que se investiguen las causas de los defectos de
calidad, y se adopten medidas apropiadas con respecto a los productos
defectuosos para prevenir que los defectos se repitan
3. SANITIZACION E HIGIENE
3.1 Cada uno de los aspectos de la fabricación de
productos farmacéuticos debe ir acompañado de un elevado nivel de saneamiento e
higiene, el cual debe abarcar al personal, locales, equipos y aparatos,
materiales y recipientes para la producción, productos de limpieza y
desinfección y todo aquello que puede ser fuente de contaminación del producto.
Todas las posibles fuentes de contaminación deben ser eliminadas mediante un
programa amplio de saneamiento e higiene.
4. CALIFICACION Y VALIDACION
4.1 De acuerdo con las BPF, cada compañía farmacéutica
debe identificar que trabajos de calificación y validación son requeridos para
probar que los aspectos críticos de sus operaciones particulares son
controlados. Debe emplearse un enfoque de análisis de riesgo para determinar el
ámbito y la amplitud de la validación. (Ver Anexo I y II).
4.2 Los elementos claves de un programa de calificación y
validación de una compañía, deben estar claramente definidos y documentados en
un plan maestro de validación.
4.3 La calificación y validación debe establecer y proveer
evidencia documentada que:
(a) los locales, sistemas de soporte, equipamiento y
procesos han sido diseñados de acuerdo con los requerimientos para las BPF
(calificación de diseño o DQ);
(b) los locales, sistemas de soporte y equipamiento han
sido construidos e instalados en cumplimiento con las especificaciones de su
diseño (calificación de instalación o IQ);
(c) los locales, sistemas de soporte y equipamiento operan
de acuerdo con las especificaciones de su diseño (calificación operacional u
OQ);
(d) un proceso específico producirá consistentemente un
producto con sus especificaciones predeterminadas y atributos de calidad
(validación de proceso, también llamado calificación de funcionamiento o PQ).
4.4 Cualquier aspecto de operación, incluyendo cambios
significativos en los locales, instalaciones, equipamiento o procesos, los
cuales puedan afectar la calidad del producto directa o indirectamente, debe
estar calificado y validado.
4.5 La calificación y validación no deben considerarse
excluyentes. Deben basarse en una revisión anual y se debe seguir un programa
continuo para su implementación.
4.6 El compromiso de mantener un estado de validación
continua debe estar especificado en la documentación relevante de la compañía,
como el manual de calidad o un plan maestro de validación
4.7 La responsabilidad de llevar a cabo la validación debe
estar claramente definida.
4.8 Estudios de validación son una parte esencial de las
BPF y deben ser conducidos de acuerdo con protocolos predefinidos y aprobados.
4.9 Se debe preparar y guardar un reporte escrito,
resumiendo los resultados registrados y las conclusiones alcanzadas.
4.10 Los procesos y procedimientos deben estar
establecidos en base a los resultados de la validación realizada.
4.11 Es de importancia crítica prestar particular atención
a la validación de métodos analíticos de ensayo, sistemas automatizados y
procedimientos de limpieza.
5. RECLAMOS
5.1 Principio. Todos los reclamos y otras informaciones
relacionadas con productos potencialmente defectuosos deben estar cuidadosamente
examinadas de acuerdo con procedimientos operativos normatizados y se deben
tomar las acciones correctivas correspondientes.
5.2 Debe ser designada una persona que se responsabilice
de atender todos los reclamos y de decidir qué medidas deben adoptarse,
juntamente con personal suficiente para asistirle en esa tarea. Si esta persona
es diferente de la persona autorizada, entonces ésta debe ser informada acerca
de todo reclamo, investigación, o retiro de productos.
5.3 Se debe contar con procedimientos operativos
normatizados que describan las medidas que deban adoptarse, incluyendo la
necesidad de que un producto sea retirado, en caso de reclamo referente a
posibles defectos del mismo.
5.4 Se debe prestar especial atención para establecer si
una queja fue causada por falsificación.
5.5 Cualquier queja concerniente a un producto defectuoso
debe ser registrada con todos los detalles originales e investigada
minuciosamente. La persona responsable de control de calidad debe estar
involucrada normalmente en la revisión de dichas investigaciones.
5.6 Si se descubre o sospecha de un producto defectuoso en
un lote de producción, debe considerarse la necesidad de constatar otras
elaboraciones, para determinar si estas también han sido afectadas.
5.7 La acción apropiada a seguir debe tomarse después de
una investigación y evaluación del reclamo, posiblemente esta acción incluya
retiro de productos del mercado.
5.8 Todas las decisiones y medidas tomadas como resultado
de un reclamo deben ser registradas y referenciadas a los registros de
producción correspondientes.
5.9 Los registros de reclamos deben ser revisados
regularmente para cualquier identificación de problemas específicos o
recurrentes que requieran atención y que pueden justificar el retiro de
productos de mercado.
5.10 La Autoridad Sanitaria Nacional debe ser informada si un elaborador está considerando acciones a seguir, teniendo en cuenta
cualquier problema serio que afecte la calidad de un producto.
6. RETIRO DE PRODUCTOS
6.1 Principio. Debe existir un sistema para retirar del
mercado en forma rápida y efectiva un producto cuando éste tenga un defecto o
exista sospecha de ello.
6.2 La persona autorizada debe ser responsable de la
ejecución y coordinación de retiros. Esta debe tener personal suficiente para
tratar todos los aspectos de los retiros con el grado apropiado de urgencia.
6.3 Deben establecerse procedimientos operativos
normatizados, los cuales serán revisados y actualizados regularmente, para la
organización de cualquier actividad de retiro. Las operaciones de retiro deben
ser capaces de iniciarse prontamente en el nivel de la cadena de distribución
donde se encuentre el producto.
6.4 Debe ser incluida una instrucción en los
procedimientos operativos normatizados para almacenar productos retirados en un
área segregada y segura mientras se decide su destino.
6.5 Todas las autoridades competentes de todos los países
a los cuales un producto dado ha sido distribuido, deben ser informadas
prontamente de cualquier intención para retirarlo porque es, o se sospecha de
ser, defectuoso.
6.6 Los registros de distribución deben estar rápidamente
disponibles para la persona autorizada y ellos deben contener suficiente
información sobre mayoristas y clientes abastecidos directamente (incluyendo,
para los productos de exportación, aquellos quienes han recibido muestras para
pruebas clínicas y médicas) para permitir un retiro efectivo.
6.7 El progreso del proceso de retiro del producto del
mercado, debe ser monitoreado y registrado. Los registros deben incluir el
destino del producto. Se debe editar un reporte final que incluya una
conciliación entre las cantidades de los productos entregados y devueltos.
6.8 La efectividad de los planes para los retiros debe ser
controlada y evaluada periódicamente.
7. CONTRATO DE PRODUCCION Y ANALISIS
7.1 Principio. El contrato de producción y análisis (sólo
los autorizados por la Autoridad Sanitaria Nacional) debe estar correctamente
definido, acordado y controlado para evitar malos entendidos que puedan influir
en un producto, trabajo o análisis de calidad no satisfactorio.
Generalidades
7.2 Todas las gestiones para el contrato de fabricación y
análisis incluyendo cualquier cambio propuesto en lo técnico u otros arreglos,
deben estar de acuerdo con la autorización de comercialización para el producto
concerniente.
7.3 El contrato debe permitir que el contratante someta a
auditorías las instalaciones del contratado.
7.4 En el caso del análisis por contrato, la aprobación
final para su liberación debe estar dada por la persona autorizada.
Contratante
7.5 El contratante es responsable de evaluar la
competencia del contratado para llevar a cabo el trabajo o ensayos requeridos
exitosamente, para la aprobación de actividades por contrato y para asegurarse
mediante el contrato que los principios de las BPF descriptos en esta guía sean
cumplidos.
7.6 El contratante debe proveer al contratado toda la
información necesaria para llevar a cabo correctamente las operaciones
contratadas conforme a la autorización de comercialización y cualquier otro
requisito legal. El contratante debe asegurar que el contratado tiene pleno
conocimiento de todos los problemas relacionados con el producto, trabajo o
ensayos que podrían causar un peligro a las instalaciones, equipamiento,
personal y otros materiales o productos.
7.7 El contratante debe asegurar que todos los productos
procesados y materiales entregados por el contratado cumplan con las
especificaciones establecidas en la autorización de comercialización y que el
producto ha sido liberado por la persona autorizada.
Contratado
7.8 El contratado debe tener instalaciones, equipamiento,
conocimiento y experiencia adecuada y personal competente para llevar a cabo
satisfactoriamente el trabajo ordenado por el contratado. El contrato de
elaboración debe ser tomado solamente por un elaborador que cuente con
autorización de elaboración de la Autoridad Sanitaria Nacional.
7.9 El contratado no debe pasar a un tercero ninguna parte
de los trabajos confiados.
7.10 El contratado debe evitar cualquier actividad que
pueda afectar adversamente la calidad de un producto fabricado y/o analizado
para el contratante.
Contrato
7.11 Debe haber un contrato escrito entre el contratante y
el contratado, el cual establezca claramente las responsabilidades de cada
parte.
7.12 El contrato debe establecer la forma en la cual la
persona autorizada, libera cada lote de producto para la venta o extiende un
certificado de análisis, ésta ejerce su responsabilidad total y asegura que
cada lote ha sido fabricado y controlado en cumplimiento con los requisitos de
la autorización de comercialización.
7.13 Los aspectos técnicos del contrato deben ser escritos
en forma apropiada por personas competentes con conocimiento en tecnología
farmacéutica, análisis y las BPF.
7.14 Todos los arreglos para la producción y análisis
deben estar de acuerdo con la autorización de comercialización y lo acordado
entre ambas partes.
7.15 El contrato debe describir claramente la
responsabilidad del contratante en la compra, control y liberación de
materiales y tiene la responsabilidad de muestrear y analizar productos
intermedios y terminados.
7.16 Los registros de producción, análisis, distribución y
muestras de referencia deben ser guardados por el contratante. Cualquier
registro relevante para investigar la calidad de un producto en caso de
reclamos o sospecha de defecto debe ser accesible y estar especificado en los
procedimientos de reclamo o defecto del contratante.
7.17 El contrato debe describir el manejo de materias
primas, intermedios, productos a granel y productos terminados, si son
rechazados. También debe describir el procedimiento a seguir si el análisis
demuestra que el producto controlado debe ser rechazado.
8. AUTOINSPECCION Y AUDITORIAS DE CALIDAD
8.1 Principio. El objetivo de la autoinspección es evaluar
el cumplimiento del fabricante con la BPF en todos los aspectos de producción y
control de calidad.
El programa de autoinspección debe ser diseñado para
detectar cualquier inconveniente en la implementación de las BPF y para
recomendar las acciones correctivas necesarias. Las autoinspecciones deben ser
realizadas rutinariamente y tal vez, realizarse además en ocasiones especiales,
por ejemplo: en casos de retiros de productos o repetidos rechazos, o cuando se
anuncia una inspección por las autoridades sanitarias. El equipo responsable
para la autoinspección debe consistir en personal que pueda evaluar la
implementación de las BPF objetivamente. Se deben implementar todas las
recomendaciones para la toma de acciones correctivas. El procedimiento para la
autoinspección debe ser documentado y debe existir un programa de continuidad
efectiva.
Elementos para la autoinspección
8.2 Instrucciones escritas para la autoinspección deben
ser establecidas para proveer un estándar de requisitos mínimos y uniformes.
Estos pueden incluir cuestionarios sobre requerimientos de las BPF que cubran
al menos los siguientes ítems:
(a) personal;
(b) instalaciones que incluyan las destinadas al personal;
(c) mantenimiento de edificios y equipamiento;
(d) almacenamiento de materias primas y productos
terminados;
(e) equipamiento;
(f) producción y controles en proceso;
(g) control de calidad;
(h) documentación;
(i) sanitización e higiene;
(j) programas de validación y revalidación;
(k) calibración de instrumentos o sistemas de medidas;
(l) procedimientos de retiro del mercado;
(m) manejo de reclamos;
(n) control de rótulos;
(o) resultado de autoinspecciones previas y cualquier
acción correctiva tomada.
Equipo de autoinspección
8.3 La dirección de la empresa debe designar un equipo de
autoinspección formado por personas expertas en sus respectivos campos y
conocedoras de las BPF. Pueden integrar dicho equipo personas de la compañía o
personas ajenas a ella.
Frecuencia de autoinspección
8.4 La frecuencia debe ser establecida en un procedimiento
operativo normatizado. La autoinspección debe realizarse, por lo menos, una vez
al año.
Reporte de autoinspección
8.5 Al finalizar se debe hacer un reporte que incluya:
(a) resultados de autoinspecciones;
(b) evaluación y conclusiones;
(c) acciones correctivas recomendadas.
Acciones de seguimiento
8.6 Debe existir un programa de seguimiento efectivo. La Dirección de la compañía debe evaluar tanto el reporte de la autoinspección como las acciones
correctivas recomendadas.
Auditorías de calidad
8.7 Podría ser conveniente complementar la autoinspección
con una auditoría de calidad. Una auditoría de calidad consiste en un examen y
evaluación de toda la parte del sistema de calidad con un propósito específico
de mejorarlo. Una auditoría de calidad es conducida, generalmente por
especialistas externos o independientes, o un equipo designado por la gerencia
para este propósito. Tal auditoría también puede extenderse a proveedores y
contratados.
Auditoría de proveedores y aprobación
8.8 La persona responsable de garantía de calidad debe
tener la responsabilidad, junto con otros departamentos relevantes, de la
aprobación de proveedores, quienes deben proveer confiablemente las materias
primas y materiales de acondicionamiento que reúnen las especificaciones
establecidas y aquellos servicios que afecten directamente o puedan comprometer
la calidad del producto y/o las BPF.
8.9 Los proveedores deben ser evaluados antes de ser
aprobados e incluidos en el registro de proveedores aprobados. Para su
evaluación se debe tener en cuenta el historial del proveedor y la naturaleza
de los materiales y/o servicios a ser provistos. Si se requiere una auditoría,
en esta se debe determinar la capacidad del proveedor para cumplir con los
estándares de BPF. (Ver Anexo V para la auditoría y calificación de proveedores
de materiales de acondicionamiento).
9. PERSONAL
9.1 Principio. El establecimiento y mantenimiento de un
sistema de Garantía de Calidad adecuado, las BPF y Control de Calidad de
productos farmacéuticos e ingredientes activos dependen de los recursos
humanos. Por esta razón, debe haber suficiente personal calificado para llevar
a cabo las tareas para las cuales el elaborador es responsable. Las
responsabilidades individuales deben estar claramente definidas y entendidas
por las personas concernientes y registradas como descripciones escritas.
Generalidades
9.2 El elaborador debe tener un número adecuado de
personal con las calificaciones y experiencias prácticas necesarias y con sus
responsabilidades definidas.
9.3 Todo personal responsable debe tener obligaciones
específicas registradas en descripciones escritas y autoridad adecuada para
llevar a cabo sus responsabilidades. Sus obligaciones pueden ser delegadas a
jefes designados con un nivel satisfactorio de calificación. No deben existir
vacíos o superposiciones en las responsabilidades del personal concerniente con
la aplicación de las BPF. El elaborador debe contar con un organigrama.
9.4 Todo el personal debe conocer los principios de las
BPF con relación a sus trabajos y recibir entrenamiento inicial y continuo,
incluyendo instrucciones de higiene relevantes a sus necesidades. Todo el
personal debe estar motivado para apoyar el establecimiento y mantenimiento de
estándares de alta calidad.
9.5 Se deben tomar medidas para prevenir la entrada a
personas no autorizadas a las áreas de producción, depósito y control de
calidad. El personal que no trabaje en estas áreas no debe usarlas como
pasillo.
Personal clave
9.6 El personal clave incluye al jefe de producción, al
jefe de control de calidad y la persona autorizada. Los puestos claves deben
ser ocupados por personal de tiempo completo. Los jefes de producción y control
de calidad deben ser independientes uno del otro. En grandes organizaciones
puede ser necesario delegar algunas de las funciones, sin embargo no se puede
delegar la responsabilidad.
9.7 El personal clave responsable de supervisar la
fabricación y control de calidad de productos farmacéuticos, debe poseer las
cualidades de una educación científica y experiencia práctica requerida por la Legislación Nacional.
9.8 El jefe de producción y el de control de calidad
generalmente tienen algunas responsabilidades compartidas, o conjuntamente
ejercidas relacionadas con la calidad. Estas pueden incluir, dependiendo de las
regulaciones nacionales:
(a) la autorización de procedimientos operativos
normatizados y otros documentos, incluyendo las modificaciones;
(b) monitoreo y control del medio ambiente de fabricación;
(c) higiene de la planta;
(d) procesos de validación y calibración de aparatos
analíticos;
(e) entrenamiento incluyendo la aplicación y principios de
garantía de calidad;
(f) aprobación y monitoreo de proveedores de materiales;
(g) aprobación y monitoreo del contrato de los
fabricantes;
(h) establecimiento y vigilancia de condiciones de
almacenamiento para materiales y productos;
(i) funcionamiento y evaluación de controles en proceso;
(j) archivo de registros;
(k) monitoreo del cumplimiento con los requisitos de las
BPF;
(l) inspección, investigación y toma de muestras para
monitorear los factores que puedan afectar la calidad del producto.
9.9 El jefe de producción generalmente tiene las siguientes
responsabilidades:
(a) asegurar que los productos se elaboren y almacenen de
acuerdo a la documentación apropiada para obtener la calidad requerida;
(b) aprobar las instrucciones relacionadas con las
operaciones de producción, incluyendo los controles en proceso, y asegurar su
estricta implementación;
(c) asegurar que los registros de producción se evalúen y
firmen por la persona designada;
(d) controlar el mantenimiento del departamento, locales y
equipamiento;
(e) asegurar que las validaciones de los procesos y las
calibraciones de los equipos de control se realicen, y se registren los
reportes;
(f) asegurar que se lleve a cabo el entrenamiento del
personal de producción, inicial y continuo, requerido y que se adapte de
acuerdo a las necesidades.
9.10 El jefe de control de calidad generalmente tiene las
siguientes responsabilidades:
(a) aprobar o rechazar materias primas, materiales de
acondicionamiento y productos intermedios, a granel y terminados, en relación
con sus especificaciones;
(b) evaluar los registros de producción;
(c) asegurar que se lleven a cabo todas las evaluaciones
necesarias;
(d) aprobar instrucciones de muestreos, especificaciones,
métodos de análisis y otros procedimientos de control de calidad;
(e) aprobar y monitorear los análisis llevados a cabo por
contrato;
(f) controlar el mantenimiento del departamento, locales y
equipamiento;
(g) asegurar que las validaciones apropiadas incluyendo
las de procedimientos analíticos y que se lleven a cabo calibraciones del equipamiento
de control;
(h) asegurar que se lleve a cabo el entrenamiento del
personal de control de calidad, inicial y continuo, requerido y que se adapte
de acuerdo a las necesidades.
Otras obligaciones de control de calidad se resumen en las
secciones 17.3 y 17.4.
9.11 La persona autorizada es responsable de cumplir con
los requerimientos técnicos y regulatorios relacionados a la calidad de los
productos terminados y la aprobación para la liberación de dichos productos
para la venta.
9.12 La persona autorizada estará involucrada en otras
actividades incluyendo las siguientes:
(a) implementación (y cuando sea necesario,
establecimiento) del sistema de calidad;
(b) participación en el desarrollo del manual de calidad
de la compañía;
(c) supervisión de auditorías internas regulares o
autoinspecciones;
(d) vigilancia del departamento de control de calidad;
(e) participación de auditorías externas;
(f) participación en programas de validación.
9.13- La función de la aprobación para la liberación de un
lote o producto terminado, pueden ser delegadas a una persona designada con
cualidades y experiencia apropiadas, quien liberará el producto de acuerdo con
un procedimiento operativo normatizado. Esto normalmente se realiza bajo
garantía de calidad por medio de una revisión del lote.
9.14- La persona responsable para aprobar un lote para su
liberación debe siempre asegurar que sean reunidos los siguientes
requerimientos:
(a) se han reunido los requerimientos de la autorización
de comercialización y de elaboración del producto, en el lote correspondiente;
(b) se han seguido los principios y guías de las BPF;
(c) que los principales procesos de control y elaboración
han sido validados;
(d) todos los controles y evaluaciones necesarios han sido
llevados a cabo, y las condiciones de producción y registros de producción han
sido tenidos en cuenta;
(e) cualquier cambio planeado o desviaciones en la
elaboración o control de calidad han sido notificados de acuerdo con un sistema
de reporte definido antes que cualquier producto sea liberado;
(f) que para cubrir los cambios planeados y desviaciones
se han llevado a cabo muestreo, inspección, pruebas y controles adicionales;
(g) toda la documentación necesaria de producción y
control de calidad ha sido completada y endosada por los supervisores
entrenados en las disciplinas necesarias;
(h) las auditorías apropiadas, autoinspecciones y
verificación de programas se han llevado a cabo por el personal entrenado y
experimentado;
(i) la aprobación ha sido emitida por el jefe de control
de calidad;
(j) que todos los factores relevantes han sido
considerados, incluyendo cualquiera que no esté especialmente asociado con la
salida directa del lote de producción bajo revisión (ej: subdivisión de
rendimiento del lote, factores asociados con la dirección de producción
continua)
10. ENTRENAMIENTO
10.1 El elaborador debe proveer entrenamiento acorde a un
programa escrito para todo el personal que tenga tareas en áreas productivas o
dentro de los laboratorios de control de calidad (incluyendo los técnicos,
personal de mantenimiento y de limpieza) y para otro personal que lo requiera.
10.2 Aparte de un entrenamiento básico teórico y práctico
de BPF, el personal recientemente incorporado debe recibir el entrenamiento
apropiado a las tareas asignadas a ellos. También se debe dar entrenamiento
continuo y determinar periódicamente su efectividad práctica. Se debe disponer
de un programa de entrenamiento aprobado. Los registros de entrenamiento deben
ser guardados.
10.3 Se debe dar un entrenamiento específico al personal
que trabaja en áreas donde exista riesgo de contaminación, ej.: áreas limpias o
áreas donde se manipulan activos altamente tóxicos, infecciosos o materiales
sensibilizantes.
10.4 El concepto de garantía de la calidad y todas las
medidas que ayudan a su entendimiento e implementación, se deben discutir
completamente durante las sesiones de entrenamiento.
10.5 Los visitantes o personal no entrenado deben
preferentemente no tener acceso a las áreas productivas y de control de
calidad. Si esto es inevitable, se les debe dar la información pertinente por
adelantado (particularmente acerca de higiene personal) y la ropa protectora
indicada. Se los debe supervisar atentamente.
10.6 El personal contratado y consultado debe ser
calificado para el servicio que ellos proveen. Esta evidencia debe ser incluida
en los registros de entrenamiento.
11. HIGIENE PERSONAL
11.1 Todo el personal, antes y durante su empleo debe
realizarse exámenes de salud. El personal que realiza inspecciones visuales
debe realizarse exámenes periódicos pertinentes.
11.2 Todo el personal debe estar entrenado en prácticas de
higiene personal. Un alto nivel de higiene personal debe ser observado por
todos aquellos que se hallan relacionados con procesos de elaboración. En
particular, el personal debe estar instruido para lavarse las manos antes de
entrar a las áreas productivas. Las señalizaciones para este efecto deben ser
claras y visibles, observando las instrucciones correspondientes.
11.3 No se debe permitir manejar materias primas,
materiales de acondicionamiento, materiales en proceso o productos medicinales
a aquellas personas que muestren en algún momento la apariencia de tener una
enfermedad evidente o lesiones abiertas que puedan afectar adversamente la
calidad de los productos, hasta que dichas condiciones no sean consideradas un
riesgo.
11.4 Todos los empleados deben estar instruidos y
estimulados a reportar inmediatamente a su supervisor cualquier condición
(relacionada con la planta, equipamiento o personal) que ellos consideren pueda
afectar adversamente la calidad de los productos.
11.5 Todo el personal debe contar con guantes apropiados a
fin de evitar el contacto directo entre las manos del operario y las materias
primas, materiales de acondicionamiento primario y productos intermedios o a
granel.
11.6 Para asegurar la protección de los productos frente a
la contaminación, el personal debe vestir ropa limpia apropiada a las tareas
que desempeña, incluyendo cubre cabello apropiado. Las ropas usadas, si son
re-utilizables, deben guardarse en contenedores o envases separados y cerrados
hasta que sean apropiadamente lavadas, y si es necesario, desinfectadas o
esterilizadas.
11.7 No se permite fumar, comer, beber, mascar, y mantener
plantas, comida, bebida, materiales para fumar y medicamentos personales en
áreas de elaboración, laboratorios y depósitos o en cualquier otra área donde
puedan influenciar adversamente la calidad del producto.
11.8 Los procedimientos de higiene personal incluyendo el
uso de ropa de protección deben ser aplicados a todo el personal que ingresa a
las áreas de producción, tanto si son empleados temporarios, permanentes o no
empleados, ej. Empleados contratados, visitantes, gerentes de categoría e
inspectores.
12. LOCALES
12.1 Principio. Los locales deben estar ubicados,
diseñados, destinados, construidos, adaptados y mantenidos para que los mismos
sean aptos para realizar las operaciones.
Generalidades
12.2 La distribución y el diseño de los locales debe apuntar
a disminuir el riesgo de errores y permitir una limpieza y mantenimiento
efectivos para evitar la contaminación, contaminación cruzada, acumulación de
polvo o suciedad, y en general, cualquier efecto adverso sobre la calidad de
los productos.
12.3 Donde el polvo es generado (ej. durante el muestreo,
pesada, mezclado y operaciones de proceso, envasado de polvos), se deben tomar
medidas para evitar la contaminación cruzada y facilitar la limpieza.
12.4 Los locales deben situarse en un ambiente que, considerado
junto con medidas para proteger el proceso de elaboración, presente un riesgo
mínimo para causar cualquier contaminación de materiales o productos.
12.5 Los locales usados para la elaboración de producto
terminado deben ser convenientemente diseñados y construidos para facilitar la
correcta sanitización.
12.6 Los locales deben ser cuidadosamente mantenidos, y se
debe asegurar que las operaciones de reparación y mantenimiento, no presenten
ningún peligro a la calidad de los productos.
12.7 Los locales deben ser limpiados, y donde sea
aplicable desinfectados, de acuerdo a procedimientos operativos normatizados.
Los registros deben ser mantenidos.
12.8 El suministro eléctrico, iluminación, temperatura,
humedad y ventilación deben ser apropiados tal que no influyan negativamente,
directa o indirectamente, ni a los productos farmacéuticos durante su
elaboración y almacenamiento, ni el correcto funcionamiento del equipamiento.
12.9 Los locales deben ser diseñados y equipados para
proporcionar la máxima protección contra la entrada de insectos, pájaros y
otros animales. Debe haber un procedimiento operativo normatizado para el
control de roedores y plagas.
12.10 Los locales deben ser diseñados para garantizar un
flujo lógico de materiales y del personal.
Areas Auxiliares
12.11 Las salas de descanso y refrigerio deben estar
separadas de las áreas de elaboración y control.
12.12 Las instalaciones para el cambio y almacenado de
ropa y para propósitos de lavado y sanitarios deben ser fácilmente accesibles y
apropiadas para el número de usuarios. Los cuartos de baño no deben estar
comunicados directamente con las áreas productivas y de depósito.
12.13 Los talleres de mantenimiento deben estar separados
de las áreas de producción. Siempre que las partes y herramientas sean
almacenadas en las áreas de producción, estas deben ser guardadas en cuartos o
armarios reservados para tal uso.
12.14 Los alojamientos de animales (bioterios) deben estar
bien aislados de otras áreas, con entradas (acceso de animales) e instalaciones
de manejo de aire independientes.
Areas de depósito
12.15 Las áreas de depósito deben ser de capacidad
suficiente para permitir un almacenamiento ordenado de varias categorías de
materiales y productos con correcta separación y segregación: materias primas,
materiales de empaque, productos intermedios, graneles y producto terminado,
productos en cuarentena y productos aprobados, rechazados, de devolución o
retiros del mercado.
12.16 Las áreas de depósito deben ser diseñadas o
adaptadas para asegurar buenas condiciones de almacenamiento. En particular,
deben estar limpias, secas y suficientemente iluminadas y mantenidas a
temperaturas compatibles con los elementos almacenados. Donde sean requeridas
condiciones especiales de almacenamiento (ej.: temperatura y humedad) estas
deben ser provistas, controladas, monitoreadas y registradas.
12.17 Las zonas de recepción y expedición deben estar
separadas, y además deben proteger a los materiales y productos de las
condiciones ambientales externas. Las áreas de recepción deben estar diseñadas
y equipadas para, cuando sea necesario, permitir la limpieza de los
contenedores de los materiales entrantes antes de su almacenamiento.
12.18 Cuando el estado "en cuarentena" se
asegure mediante el almacenamiento en áreas separadas, estas áreas deben estar
claramente indicadas y su acceso restringido a personal autorizado. Cualquier
sistema reemplazante de la cuarentena física debe brindar una seguridad
equivalente.
12.19 Se debe mantener segregado el almacenamiento de
materiales o productos rechazados, retirados del mercado o devoluciones.
12.20 Los materiales altamente activos y radioactivos,
psicotrópicos y estupefacientes, otras drogas peligrosas y sustancias que presenten
especial riesgo de abuso, fuego o explosión, deben ser almacenadas en áreas
seguras.
12.21 Los materiales de acondicionamiento impresos son
considerados críticos y se debe prestar especial atención.
12.22 Normalmente debe haber un área de muestreo separada
para las materias primas.
Areas de pesada
12.23 La pesada de materias primas y la estimación del
rendimiento por pesada deben ser realizadas en áreas de pesada separadas
destinadas para tal fin, por ejemplo, provistas para el control de polvo. Tales
áreas pueden ser parte de uno de los depósitos o áreas de producción.
Areas de Producción
12.24 Para minimizar el riesgo de un peligro médico serio
debido a la contaminación cruzada, se debe disponer de áreas dedicadas para la
elaboración de productos farmacéuticos particulares como aquellos conteniendo
materiales altamente sensibilizantes (ej: Penicilinas), preparaciones
biológicas (ej: microorganismos vivos) o materiales altamente activos tales
como hormonas, algunos antibióticos y citotóxicos. En casos excepcionales puede
ser aceptado el principio del "trabajo en campaña" (separación en el
tiempo) en las mismas instalaciones, siempre que se tomen las precauciones
específicas y sean hechas las validaciones necesarias (incluyendo validación de
limpieza). La elaboración de productos tóxicos tales como, pesticidas y
herbicidas, no se deben permitir en los locales usados para la elaboración de
productos farmacéuticos.
12.25 Los locales deben ser distribuidos de forma tal que
permitan que la elaboración se lleve a cabo en áreas conectadas en un orden
lógico, que corresponda a la secuencia de las operaciones y a los niveles
requeridos de limpieza.
12.26 La adecuación del trabajo y del lugar de
almacenamiento en proceso, debe permitir la ubicación lógica y ordenada del
equipamiento y materiales para minimizar el riesgo de la confusión entre
diferentes productos farmacéuticos o sus componentes, para evitar la
contaminación cruzada, y minimizar el riesgo de la omisión o mala aplicación de
cualquier etapa de elaboración o control.
12.27 Donde las materias primas y los materiales de
acondicionamiento primario, y los productos intermedios o a granel son
expuestos al ambiente, las superficies interiores (paredes, pisos y techos)
deben ser lisas, sin grietas, ni empalmes abiertos, no deben liberar partículas
de material, y deben facilitar la limpieza y, en caso de ser necesario, la
fácil y eficaz desinfección.
12.28 Las cañerías, los montajes de luz, los puntos de
ventilación y otros servicios deben estar diseñados y situados para evitar la
creación de hendiduras que sean dificultosas para limpiar. Si es posible, para
el propósito de su mantenimiento, éstos deben ser accesibles por fuera de las
áreas de elaboración.
12.29 Los desagües deben ser de las medidas adecuadas,
diseñados y equipados para prevenir el reflujo. Los canales abiertos deben ser
evitados cuando sea posible, pero si estos son necesarios deben ser poco
profundos para facilitar la limpieza y desinfección.
12.30 Las áreas de producción deben ser ventiladas
efectivamente con equipos de control de aire (incluyendo filtración del aire al
nivel suficiente para prevenir la contaminación y la contaminación cruzada,
como así también el control de temperatura y, donde sea necesario, humedad),
apropiados para el manipuleo de productos, para las operaciones emprendidas y
para el medio ambiente externo. Estas áreas deben ser monitoreadas regularmente
durante los períodos de producción y no producción, para asegurar el
cumplimiento con sus especificaciones de diseño.
12.31 Los locales de acondicionamiento de productos
farmacéuticos deben estar diseñados específicamente y con una distribución
adecuada para evitar la mezcla o contaminación cruzada.
12.32 Las áreas de producción deben estar bien iluminadas,
particularmente cuando se lleven a cabo controles visuales en línea.
Areas de Control de Calidad
12.33 Los laboratorios de control de calidad deben estar
separados de las áreas de producción. Las áreas donde se realicen ensayos
biológicos, microbiológicos o con radioisótopos, deben estar separadas entre
sí.
12.34 Los laboratorios de control de calidad deben estar
diseñados para la demanda de las operaciones que van a llevarse a cabo en
ellos. Debe ser concedido suficiente espacio para evitar las mezclas y la
contaminación cruzada. Allí, debe haber un espacio adecuado para almacenar
muestras, sustancias y materiales de referencia (si fuese necesario con
refrigeración), solventes, reactivos y registros.
12.35 En el diseño de los laboratorios se debe tomar en
cuenta la adecuación de los materiales de construcción, prevención de vapores y
ventilación. Allí debe estar separado el suministro de aire del de producción.
Es necesario separar las unidades manejadoras de aire y otros abastecimientos
para laboratorios biológicos, microbiológicos y de radioisótopos.
12.36 Para los instrumentos, podría ser necesario un
cuarto separado para protegerlos contra interferencias eléctricas, vibraciones,
contacto con excesiva humedad y otros factores externos, o donde sea necesario
aislar los instrumentos.
13. EQUIPAMIENTO
13.1 El equipamiento debe estar localizado, diseñado,
construido, adaptado y mantenido para satisfacer las operaciones que se lleven
a cabo. La distribución y la disposición del equipamiento debe apuntar a
minimizar el riesgo de errores y permitir la limpieza efectiva y mantenimiento
para evitar la contaminación cruzada, acumulación de polvo o suciedad, y en
general, cualquier efecto adverso sobre la calidad de los productos.
13.2 Los equipos deben estar instalados de una manera tal
que se minimice cualquier riesgo de error o de contaminación.
13.3 En las tuberías fijas debe indicarse claramente el
contenido y, donde sea aplicable, la dirección del flujo.
13.4 Todos los servicios de cañerías y dispositivos deben
estar adecuadamente marcados y prestar especial atención para la provisión de
conexiones no intercambiables o adaptadores para gases o líquidos peligrosos.
13.5 Deben estar disponibles balanzas y otros equipos de
medición, calibrados con una frecuencia programada, de rango y precisión
adecuados para operaciones de producción y control.
13.6 Los equipos de producción deben limpiarse a fondo con
una frecuencia programada.
13.7 Los equipos y los instrumentos de laboratorio deben
ser adecuados a los métodos de prueba emprendidos.
13.8 Los equipos de limpieza, lavado y secado deben ser
elegidos y utilizados tal que no sean una fuente de contaminación.
13.9 El equipo de producción no debe presentar ningún
peligro para los productos. Las partes del equipo de producción que están en
contacto con el producto no deben ser reactivas, aditivas, o absorbentes a un
grado que afecte la calidad del producto.
13.10 Los equipos defectuosos se deben quitar de las áreas
productivas y de control de calidad. Si esto no es posible, deben ser
claramente identificados como defectuosos para evitar su uso.
13.11 Los equipos cerrados deben utilizarse siempre que
sea apropiado. Donde se utilice un equipo abierto o cuando se proceda a la apertura
del equipo, se deben tomar las precauciones para reducir al mínimo la
contaminación.
13.12 Los equipos no dedicados se deben limpiar según
procedimientos de limpieza validados entre la elaboración de diferentes
productos farmacéuticos para prevenir la contaminación cruzada.
13.13 Se deben guardar los planos actualizados de los
equipos críticos y sistemas de soporte.
14. MATERIALES
14.1 Principio. El principal objetivo de las plantas
farmacéuticas es elaborar productos finales para uso en pacientes a partir de
una combinación de materiales (desde las materias primas hasta el
acondicionamiento).
14.2 Los materiales incluyen materias primas, materiales
de acondicionamiento, gases, solventes, excipientes, reactivos y materiales de
rotulado.
Generalidades
14.3 Los materiales usados para operaciones, tales como
limpieza, lubricación de equipos y control de plagas, no deben estar en
contacto directo con los productos. Tales materiales deben ser de grado
adecuado para minimizar los riesgos en la salud (por ej. grado alimenticio).
14.4 Todos los materiales y productos terminados deben ser
puestos en cuarentena, inmediatamente después de ser recibidos o procesados
hasta que sean liberados para uso o distribución.
14.5 Todos los materiales y productos deben ser
almacenados bajo condiciones apropiadas establecidas por el elaborador y en
forma ordenada, para permitir separar partidas y rotación de stock por la regla
de "primero vence, primero sale".
14.6 El agua usada en la fabricación de productos
farmacéuticos debe ser la adecuada para el uso destinado.
Materias primas
14.7 La adquisición de materias primas es una operación
importante que debe involucrar gente con conocimiento particular y minucioso de
los productos y de los proveedores.
14.8 Las materias primas deben ser adquiridas sólo a
proveedores calificados y, cuando sea posible, directamente al elaborador. Se
recomienda que las especificaciones de las mismas sean las establecidas por el
elaborador del producto farmacéutico.
14.9 En cada envío, se debe controlar al menos la
integridad del embalaje, el sellado y la correspondencia entre la orden, el
remito y los rótulos del proveedor.
14.10 Todos los materiales ingresados deben ser
verificados para asegurar que el envío corresponde a la orden de compra. Los contenedores
deben limpiarse, cuando sea necesario, y rotulados con la información
preestablecida. Cuando se adosen rótulos adicionales al contenedor, la
información original no debe perderse.
14.11 Los daños a los contenedores y cualquier otro
problema que pueda afectar a la calidad de los materiales deben ser registrados
y reportados al departamento de control de calidad e investigados.
14.12 Si una entrega de material proviene de lotes
diferentes, cada lote debe ser considerado independientes para el muestreo,
control y liberación.
14.13 Las materias primas deben ser rotuladas en el área
de depósito. Los rótulos deben tener la siguiente información:
(a) nombre designado del producto y, cuando sea aplicable,
el código de referencia interno;
(b) número de lote dado por el proveedor y, cuando se
recibe, el control o número de lote dado por el elaborador, para documentar y
asegurar la trazabilidad;
(c) el estado del contenido (p.ej en cuarentena, en
control, liberado, rechazado, devuelto, recolectado);
(d) la fecha de vencimiento y la fecha de re-análisis.
Cuando se usan sistemas de almacenamiento, completamente
computarizados, no toda la información anterior necesita estar de forma legible
en el rótulo.
14.14 Deben existir procedimientos apropiados o medidas
para asegurar la identidad de los contenidos para cada contenedor de materias
primas. Los contenedores desde los cuales las muestras son tomadas deben ser
identificados.
14.15 Las materias primas sólo deben ser liberadas por el
departamento de control de calidad dentro de su vida útil.
14.16 Las materias primas deben ser dispensadas sólo por
las personas designadas, siguiendo un procedimiento operativo normatizado para
asegurar que los materiales correctos sean precisamente pesados o medidos
dentro de contenedores limpios y correctamente rotulados.
14.17 Cada material dispensado y el peso o volumen deben
ser independientemente verificados y registrados.
14.18 Para cada partida de producto terminado, los
materiales dispensados deben ser guardados juntos y detalladamente rotulados
como tales.
Materiales de acondicionamiento
14.19 La adquisición de material de acondicionamiento debe
ser realizada a través de proveedores calificados y auditados por el titular
del registro. El manipuleo y control de los envases primarios e impresos se
debe realizar con igual procedimiento al utilizado con las materias primas.
14.20 Se debe prestar particular atención a los materiales
de acondicionamiento impresos. Deben ser almacenados en condiciones seguras
para evitar la posibilidad de acceso a personal no autorizado. Los rollos de
rótulos se deben utilizar cuando sea posible. Los rótulos de corte y otros
materiales impresos deben ser almacenados y transportados en envases cerrados y
separados para evitar la confusión. Los materiales de acondicionamiento se
deben dispensar para su uso, exclusivamente por el personal designado,
siguiendo un procedimiento aprobado y documentado.
14.21 A cada envío o lote del material de acondicionamiento
impreso o primario se le debe dar un número de referencia o una identificación
específica.
14.22 El material de acondicionamiento primario o el
material de acondicionamiento impreso, vencido u obsoleto, debe ser destruido y
su destino registrado.
14.23 Debe ser comprobada la entrega de todos los
productos y materiales de acondicionamiento, que van a ser utilizados, al
departamento de acondicionamiento, junto con la cantidad, identidad y
conformidad con las instrucciones de acondicionamiento.
Productos intermedios y a granel
14.24 Los productos intermedios y a granel deben ser
almacenados bajo condiciones apropiadas.
14.25 Los productos intermedios y a granel adquiridos como
tales deben ser recibidos y tratados como materias primas.
Productos terminados
14.26 Los productos terminados deben ser conservados en
cuarentena hasta su liberación final, después de lo cual deben ser almacenados
como stock utilizable bajo las condiciones establecidas por el fabricante.
14.27 La evaluación del producto terminado y la
documentación necesaria para el lanzamiento de un producto para la venta se
describe en la sección 17, "Buenas Prácticas en Control de Calidad".
Materiales rechazados
14.28 Los materiales y productos rechazados se deben
identificar claramente como tales y almacenar por separado en áreas
restringidas. Deben ser devueltos a los proveedores o, cuando sea apropiado,
destruidos en un tiempo oportuno. La toma de cualquier acción debe ser aprobada
por el personal autorizado y ser registrado.
Productos retirados del mercado
14.29 Los productos retirados del mercado se deben
identificar y almacenar separadamente, en un área segura hasta que se tome una
decisión de su destino. La decisión debe ser tomada lo antes posible.
Devoluciones
14.30 Todo producto devuelto debe ser críticamente
analizado por control de calidad de acuerdo a un procedimiento operativo
normatizado. Se debe evaluar exhaustivamente el motivo de la devolución,
considerando además, la naturaleza del producto y condiciones de
almacenamiento. Los productos devueltos deben ser destruidos a menos que su
calidad sea comprobadamente satisfactoria; en tal caso puede ser considerado un
nuevo destino. Cualquier acción tomada debe ser registrada apropiadamente.
Reactivos y medios de cultivo
14.31 La recepción y la preparación de reactivos y medios
de cultivo deben ser registradas.
14.32 Los reactivos hechos en el laboratorio deben ser
preparados según procedimientos escritos y ser rotulados apropiadamente. El
rótulo debe indicar la concentración, el factor de la estandarización, la vida
útil, la fecha cuando se debería re-estandarizar y las condiciones de
almacenamiento. El rótulo debe ser firmado y fechado por la persona que prepara
el reactivo.
14.33 Los controles positivo y negativo deben ser
aplicados para verificar la aptitud de los medios de cultivo, cada vez que
estos son preparados y utilizados. El tamaño del inóculo usado en los controles
positivos debe ser el apropiado para la sensibilidad requerida.
Sustancias y Materiales de Referencia
14.34 Siempre que existan las sustancias oficiales de
referencia, éstas deben ser utilizadas.
14.35 Las sustancias oficiales de referencia se deben
utilizar solamente para el propósito descripto en la monografía.
14.36 Los Estandares preparados por el elaborador deben
ser ensayados, liberados y almacenados (Ver Anexo IV). Deben ser guardados bajo
responsabilidad de una persona designada en un área segura.
14.37 Los materiales de referencia preparados por el
elaborador o los estándares de trabajo se pueden establecer por la aplicación
de ensayos y controles apropiados en intervalos regulares para asegurar su
estandarización. (Ver Anexo IV).
14.38 Los materiales de referencia y los estándares de
trabajo deben ser rotulados correctamente con por lo menos la siguiente
información:
(a) nombre del material;
(b) serie o número de lote y número del control;
(c) fecha de la preparación;
(d) vida útil;
(e) potencia o concentración;
(f) condiciones de almacenamiento.
14.39 Todos los estándares de trabajo deben ser
inicialmente estandarizados contra una sustancia oficial de referencia, cuando
estén disponibles, y, después de eso, a intervalos regulares.
14.40 Todos los materiales de referencia deben ser almacenados
y utilizados de una manera que no afecte su calidad y conforme a procedimientos
escritos.
Materiales de desecho
14.41 Debe existir un local para el almacenamiento
apropiado y seguro de los materiales de desecho hasta su destino final.
14.42 No se debe permitir acumular los materiales de
desecho. Deben ser recolectados en recipientes adecuados para su traslado a los
puntos de retiro fuera de los edificios. Deben ser eliminados de manera segura
y sanitaria a intervalos regulares y frecuentes.
Misceláneos
14.43 Las sustancias tóxicas y los materiales inflamables
se deben almacenar en ambientes convenientemente diseñados según los requisitos
de la Legislación Nacional.
14.44 No se deben permitir que rodenticidas, insecticidas,
agentes de fumigación y materiales de sanitización contaminen el equipamiento,
las materias primas, los materiales de acondicionamiento, los materiales en
proceso o los productos terminados.
15. DOCUMENTACION
15.1 Principio. La buena documentación es una parte
esencial del sistema de aseguramiento de la calidad y, como tal, debe existir
para todos los aspectos de las BPF. Sus objetivos son definir las
especificaciones y procedimientos para todos los materiales y métodos de
fabricación y control; asegurar que todo el personal involucrado en la
fabricación sepa qué hacer y cuándo hacerlo; asegurar que las personas
autorizadas tengan toda la información necesaria para decidir la liberación o
no de un lote del producto para la venta, asegurar la existencia de evidencia
documentada, trazabilidad, y proveer registros y un informe de auditoría que
permita la investigación. Asegura la disponibilidad de la información necesaria
para la validación, revisión y análisis estadístico. El diseño y uso de los
documentos depende del fabricante. En algunos casos, algunos o la totalidad de
los documentos descriptos más adelante pueden agruparse, pero usualmente
estarán separados.
Generalidades
15.2 Los documentos deben diseñarse, prepararse, revisarse
y distribuirse con cuidado. Deben cumplir con las partes relevantes de las
autorizaciones de fabricación y comercialización.
15.3 Los documentos deben aprobarse, firmarse y fecharse
por las personas responsables apropiadas. Ningún documento debe modificarse sin
autorización y aprobación.
15.4 Los documentos no deben tener contenidos ambiguos: el
título, naturaleza y propósito deben estar claramente estipulados. Deben
redactarse en un estilo ordenado y ser fáciles de verificar. Los documentos
reproducidos deben ser claros y legibles. La reproducción de documentos de
trabajo a partir de documentos maestros no debe permitir que ningún error sea
introducido a través del proceso de reproducción.
15.5 Los documentos deben revisarse regularmente y
mantenerse actualizados. Cuando un documento ha sido revisado, debe existir un
sistema para prevenir el uso inadvertido de la versión reemplazada. Los
documentos reemplazados deben retenerse por un período de tiempo especificado.
15.6 Cuando los documentos requieran la entrada de datos,
estas entradas deben ser claras, legibles e indelebles. Debe proveerse de
suficiente espacio para tales ingresos.
15.7 Cualquier alteración hecha a un documento debe
firmarse y fecharse; la alteración debe permitir la lectura de la información
original y el motivo de la misma debe registrarse.
15.8 Los registros deben realizarse o completarse cuando
cualquier acción se lleve a cabo y de manera tal que todas las actividades
significativas implicadas en la fabricación de productos farmacéuticos sean
trazables. Los registros deben retenerse al menos por un año luego de la fecha
de vencimiento del producto terminado.
15.9 La documentación y registros que deban conservarse,
podrán ser mantenidos mediante sistemas electrónicos de procesamiento de datos
o por medios fotográficos u otro medio confiable. La fórmula maestra y los
procedimientos operativos normatizados detallados relativos al sistema en uso
deben estar disponibles y la exactitud de los registros debe verificarse. Si la
documentación se maneja mediante métodos electrónicos de procesamiento de
datos, sólo las personas autorizadas deben poder ingresar o modificar los datos
en la computadora, y debe haber un registro de los cambios y deleciones; el
acceso debe restringirse mediante claves u otros medios y el ingreso de
información crítica debe verificarse en forma independiente. Los registros de
lote almacenados electrónicamente deben protegerse mediante una copia de
seguridad o cintas magnéticas, microfilms, impresiones en papel u otros medios.
Es particularmente importante que, durante el período de retención, la
información esté rápidamente disponible.
Documentos requeridos
Rótulos
15.10 Los rótulos aplicados a envases, equipos o
instalaciones deben ser claros, sin ambigüedades y con el formato acordado por
la compañía. Además del texto, es útil en los rótulos usar colores para indicar
el estado (ej. en cuarentena, aceptado, rechazado, limpio).
15.11 Todos los productos terminados deben identificarse
mediante rotulado, tal como lo requiere la legislación nacional, conteniendo al
menos la siguiente información:
(a) el nombre del producto;
(b) una lista de los principios activos (si corresponde,
con la Denominación Común Internacional –DCI-), mostrando la cantidad de cada
uno y una declaración de los contenidos netos (ej. número de unidades de
dosificación, peso, volumen);
(c) el número de lote asignado por el fabricante;
(d) la fecha de vencimiento en una forma no codificada;
(e) condiciones especiales de almacenamiento o
precauciones en la manipulación que puedan ser necesarias;
(f) indicaciones de uso, y advertencias y precauciones que
puedan ser necesarias;
(g) el nombre y dirección del fabricante o la compañía o
persona responsable de colocar el producto en el mercado.
15.12 Para los sustancias y materiales de referencia y
estandares, el rótulo y/o documentación acompañante debe indicar la potencia o
concentración, fecha de fabricación, fecha de vencimiento, fecha en que se abre
por primera vez, condiciones de almacenamiento y número de control.
Especificaciones y procedimientos de análisis
15.13 Los procedimientos de análisis descriptos en los
documentos, y no codificados en farmacopeas internacionalmente reconocidas
deben estar validados en el contexto de las instalaciones y equipamiento
disponible antes de que sean adoptados como análisis de rutina.
15.14 Debe haber especificaciones fechadas y
apropiadamente autorizadas, que incluyan ensayos de identificación, pureza y
calidad, para materias primas y material de acondicionamiento y producto
terminado; cuando corresponda, también deben estar disponibles para productos
intermedios y a granel. Deben incluirse las especificaciones para el agua,
solventes y reactivos (p.ej: ácidos y bases) usados en la producción.
15.15 Cada especificación debe aprobarse, firmarse,
fecharse y ser mantenida por control de calidad, garantía de calidad o centro
de documentación.
15.16 Serán necesarias revisiones periódicas de las
especificaciones para cumplimentar las nuevas ediciones de la farmacopea
nacional u otros compendios oficiales.
15.17 Las farmacopeas, materiales de referencia, espectros
de referencia y otras referencias deben estar disponibles en el laboratorio de
control de calidad.
Especificaciones para materias primas y material de
acondicionamiento
15.18 Las especificaciones para las materias primas y
material de acondicionamiento impreso, deben proveer, si es aplicable, una
descripción de los materiales, que incluya:
(a) el nombre designado (si corresponde, la DCI) y la referencia de código interno;
(b) la referencia, si existe, a una monografía de
farmacopea;
(c) requisitos cualitativos y cuantitativos con los
límites de aceptación.
De acuerdo a la práctica de la compañía puede adicionarse
otra información a las especificaciones, tal como:
(a) el proveedor y el productor original de los
materiales;
(b) una muestra de los materiales impresos;
(c) directivas para muestreo y análisis, o una referencia
a los procedimientos;
(d) condiciones de almacenamiento y precauciones;
(e) el máximo período de almacenamiento antes de re - análisis.
El material de acondicionamiento debe cumplir las
especificaciones, y ser compatible con el material y/o con el producto que
contiene. El material debe examinarse para el cumplimiento de las
especificaciones, verificar presencia de defectos críticos o mayores, como
también si las marcas que lo identifican son correctas.
15.19 Los documentos que describen procedimientos de
análisis deben establecer la frecuencia requerida para el reanálisis de cada
materia prima, tal como lo determine su estabilidad.
Especificaciones para productos intermedios y a granel
15.20 Las especificaciones para productos intermedios y a
granel deben estar disponibles. Las especificaciones deben ser de similares carácter
a las especificaciones para materias primas o productos terminados.
Especificaciones para productos terminados
15.21 Las especificaciones para productos terminados deben
incluir:
(a) el nombre designado del producto y el código de
referencia, donde sea aplicable;
(b) el/los nombre/s designado/s del principio activo (si
es aplicable, con la/s DCI/s);
(c) la fórmula o una referencia a la fórmula;
(d) una descripción de la forma farmacéutica y detalles
del acondicionamiento;
(e) directivas para el muestreo y análisis o una
referencia a los procedimientos;
(f) los requerimientos cualitativos y cuantitativos, con
los límites de aceptación;
(g) las condiciones de almacenamiento y precauciones,
cuando corresponda;
(h) el período de vida útil.
Fórmula maestra
15.22 Para cada producto y tamaño de lote a elaborar debe
existir una fórmula maestra oficialmente autorizada por la Autoridad Sanitaria Nacional.
15.23 La fórmula maestra debe incluir:
(a) el nombre del producto, con un código de referencia del
producto relativo a sus especificaciones;
(b) una descripción de la forma farmacéutica, potencia del
producto y tamaño de lote;
(c) una lista de todas las materias primas a utilizarse
(si corresponde, con las DCI/s), con la cantidad de cada una, descriptas usando
los nombres designados y una referencia que sea exclusiva para la sustancia
(debe mencionarse cualquier sustancia que pueda desaparecer en el curso del
procesamiento);
(d) una declaración del rendimiento final esperado con los
límites de aceptación, y de rendimientos intermedios relevantes, donde sea
aplicable.
(e) una indicación del lugar del proceso y de los
principales equipos a ser empleados;
(f) los métodos, o la referencia a los métodos, a
utilizarse en la preparación y operación de equipamiento crítico, p.ej:
limpieza (especialmente luego de un cambio de producto), ensamble, calibración,
esterilización, uso, etc.;
(g) instrucciones detalladas paso a paso del procesamiento
(p.ej: verificación de materiales, pretratamientos, secuencia de adición de
materiales, tiempos de mezclado, temperaturas, etc.);
(h) las instrucciones para cualquier control en proceso
con sus límites;
(i) donde sea necesario, los requisitos para el
almacenamiento de los productos, incluyendo el envase, el rotulado, y cualquier
condición especial de almacenamiento;
(j) Cualquier precaución especial a adoptarse.
Instrucciones de acondicionamiento
15.24 Deben existir instrucciones oficialmente autorizadas
por la Autoridad Sanitaria Nacional para el material de acondicionamiento,
tamaño y tipo de envase.
Como mínimo debe incluir, o hacer referencia a:
(a) el nombre del producto;
(b) una descripción de su forma farmacéutica, potencia y,
cuando corresponda, método de aplicación;
(c) el tamaño del envase expresado en términos de un
número, peso o volumen del producto en el envase final;
(d) una lista completa de todos los materiales de
acondicionamiento, requeridos para un tamaño de lote estándar, incluyendo
cantidades, tamaños y tipos, con el código o número de referencia relativo a
las especificaciones para cada material de acondicionamiento;
(e) un ejemplo o reproducción, según corresponda, de los
materiales de acondicionamiento impresos y muestras, que indique el lugar donde
será colocado el número de lote y la fecha de vencimiento del producto.
(f) precauciones especiales a observarse, incluyendo un
cuidadoso examen del equipo y área de acondicionamiento a los fines de
determinar la limpieza de la línea antes y después de las operaciones de
acondicionamiento;
(g) una descripción de la operación de acondicionamiento,
incluyendo cualquier operación subsidiaria relevante, y equipamiento a
utilizarse;
(h) detalles de controles en proceso con instrucciones
para el muestreo y límites de aceptación.
Registros de procesamiento del lote
15.25 Debe guardarse un registro de procesamiento de lote
para cada lote elaborado. Debe basarse en las partes pertinentes de la fórmula
maestra aprobada que esté en vigencia. El método de preparación de tales
registros debe diseñarse de forma tal de evitar errores.
15.26 Antes de comenzar el procesamiento, debe verificarse
que los equipos y la estación de trabajo estén libres de productos previos,
documentos, o materiales no requeridos para el proceso planeado, y que los
equipos estén limpios y adecuados para el uso. Esta verificación debe
registrarse.
15.27 Durante el procesamiento, la siguiente información
debe registrarse en el momento en que cada acción se lleva a cabo, y luego de
completar el registro este debe fecharse y firmarse por la persona responsable
de las operaciones de procesamiento:
(a) nombre del producto;
(b) número de lote en elaboración;
(c) fechas y horas de comienzo, de etapas intermedias
significativas, y terminación de la producción;
(d) el nombre de la persona responsable de cada etapa de
producción;
(e) las iniciales de el/los operador/es de diferentes
pasos significativos de la producción, de la/s persona/s que verificaron cada
una de estas operaciones (p.ej: pesada);
(f) el número de lote y/o número de control analítico y la
cantidad de cada materia prima pesada;
(g) cualquier operación del proceso o evento relevante y
los equipos utilizados;
(h) los controles en proceso ejecutados, las iniciales de
la persona que los llevó a cabo, y los resultados obtenidos;
(i) la cantidad de producto obtenido en diferentes y
pertinentes etapas de la elaboración (rendimiento);
(j) notas sobre problemas especiales incluyendo detalles,
con autorización firmada para cualquier desviación de la fórmula maestra.
Registros de acondicionamiento del lote
15.28 Debe guardarse un registro del acondicionamiento del
lote para cada lote o parte de lote procesados. Debe basarse en las partes
relevantes de las instrucciones de acondicionamiento aprobadas, y el método de
preparación de tales registros debe diseñarse de manera tal de evitar errores.
15.29 Antes de comenzar cualquier operación de
acondicionamiento, debe verificarse que los equipos y la estación de trabajo
estén libres de productos previos, documentos o materiales no requeridos para
las operaciones de envasado planeado, y que los equipos estén limpios y
adecuados para el uso. Estas verificaciones deben registrarse.
15.30 La información siguiente debe registrarse en el
momento en que cada acción se lleva a cabo, y la fecha y la persona responsable
deben ser claramente identificadas mediante firma o clave electrónica:
(a) el nombre del producto, el número de lote y la
cantidad de producto a granel a acondicionarse, así también como el número de
lote y la cantidad esperada de producto terminado que se obtendrá, la cantidad
finalmente obtenida y la conciliación;
(b) la/s fecha/s y hora/s de las operaciones de
acondicionamiento;
(c) el nombre de la persona responsable de llevar a cabo
la operación de acondicionamiento;
(d) las iniciales de los operadores de diferentes etapas
significativas;
(e) las verificaciones hechas para la identidad y la
conformidad con las instrucciones de acondicionamiento, incluyendo los
resultados de los controles en proceso;
(f) detalles de las operaciones de acondicionamiento
llevadas a cabo, incluyendo referencias a los equipos y a las líneas de
acondicionamiento utilizadas, y, cuando sea necesario, las instrucciones para
mantener el producto sin acondicionar o un registro del producto devuelto que
no ha sido acondicionado al área de almacenamiento;
(g) cuando sea posible, muestras de los materiales de
acondicionamiento impresos utilizados, incluyendo ejemplares con impresión
aprobada y verificación regular (donde sea apropiado) del número de lote, fecha
de vencimiento, y cualquier sobre - impresión adicional;
(h) notas de cualquier problema especial, incluyendo
detalles de cualquier desviación de las instrucciones de acondicionamiento, con
autorización escrita de una persona apropiada;
(i) las cantidades y el número de referencia o
identificación de todos los materiales de acondicionamiento impresos y producto
a granel emitido, usado, destruido o devuelto a stock y las cantidades de
producto obtenido para permitir una adecuada conciliación.
Procedimientos operativos normatizados (PON) y registros
15.31 Los procedimientos operativos normatizados y
registros asociados de acciones tomadas o, cuando corresponda, las conclusiones
obtenidas deben, entre otros, estar disponibles para:
(a) armado de equipos y validación;
(b) aparatos analíticos y calibración;
(c) mantenimiento, limpieza y sanitización;
(d) temas relacionados con el personal incluyendo
calificación, entrenamiento, vestimenta e higiene;
(e) monitoreo ambiental;
(f) control de enfermedades;
(g) quejas;
(h) retiros del mercado
(i) devoluciones.
(j) actividades relacionadas con: depósitos, producción,
control y garantía de calidad, recursos humanos, etc.
15.32 Debe haber procedimientos operativos normatizados y
registros para la recepción de cada entrega de materia prima y material de
acondicionamiento impreso.
15.33 Los registros de las recepciones deben incluir al
menos:
(a) el nombre del material en la nota de entrega y los
envases;
(b) el nombre interno y/o el código del material si este
difiere de (a);
(c) la fecha de recepción;
(d) el nombre del proveedor y, si es posible, el nombre
del elaborador;
(e) el número de lote o referencia del elaborador;
(f) la cantidad total, y número de envases recibidos;
(g) el número de lote asignado luego de la recepción;
(h) cualquier comentario relevante (p.ej: estado de los
envases).
15.34 Debe haber procedimientos operativos normatizados
para el rotulado interno, cuarentena y almacenamiento de materias primas,
materiales de envasado y otros materiales, en forma apropiada.
15.35 Deben estar disponibles los procedimientos
operativos normatizados para cada instrumento y partes de equipos (p.ej: uso,
calibración, limpieza, mantenimiento) y dispuestos en las proximidades de los
equipos.
15.36 Deben haber procedimientos operativos normatizados
para el muestreo, que especifiquen a la/s persona/s autorizadas para tomar las
muestras.
15.37 Las instrucciones de muestreo deben incluir:
(a) el método de muestreo y el plan de muestreo;
(b) los equipos a utilizarse;
(c) cualquier precaución a observarse para evitar la
contaminación del material o cualquier deterioro de su calidad;
(d) la/s cantidad/es de muestra a tomarse;
(e) instrucciones para cualquier subdivisión requerida de
la muestra;
(f) el tipo de envase para la muestra a utilizarse, y si
son para muestreo aséptico o normal, y rotulado;
(g) cualquier precaución específica a observarse,
especialmente en consideración del muestreo de material estéril o nocivo.
15.38 Debe haber procedimientos operativos normatizados
que describan los detalles del sistema de numeración del lote, con el objetivo
de asegurar que cada lote de producto intermedio, a granel o terminado se
identifique con un número específico de lote.
15.39 Los procedimientos operativos normatizados para la
numeración del lote que se apliquen a la etapa de procesamiento o la respectiva
etapa de acondicionado deben relacionarse entre sí.
15.40 El procedimiento operativo normatizado para la
numeración debe asegurar que números de lote iguales no se utilicen para el
mismo producto.
15.41 La asignación del número de lote debe registrarse
inmediatamente. El registro debe incluir al menos la fecha de asignación,
identidad del producto y tamaño de lote.
15.42 Debe haber procedimientos operativos normatizados
para el análisis de materiales y productos en diferentes etapas de la
fabricación, describiendo los métodos y equipamiento a utilizarse. Los ensayos
llevados a cabo deben registrarse.
15.43 Los registros de análisis deben incluir al menos la
siguiente información:
(a) el nombre del material o producto y, cuando
corresponda, la forma farmacéutica;
(b) el número de lote y, donde sea apropiado, el
fabricante y/o proveedor;
(c) referencias a especificaciones relevantes y
procedimientos de análisis;
(d) resultados de ensayos, incluyendo observaciones y
cálculos, y referencia a las especificaciones (límites);
(e) fechas/s y número/s de referencia de análisis;
(f) las iniciales de las personas que llevaron a cabo el
análisis;
(g) la fecha y las iniciales de las personas que
verificaron los análisis y los cálculos, cuando corresponda;
(h) una declaración clara de la liberación o rechazo (u
otra decisión de estado) y la firma fechada de la persona responsable
designada.
15.44 Deben estar disponibles los procedimientos
operativos normatizados de liberación y rechazo de materiales y productos, y en
particular para liberación para la venta del producto terminado por una persona
autorizada.
15.45 Deben mantenerse registros de la distribución de
cada lote de un producto para, por ejemplo, facilitar el retiro del mercado del
lote si es necesario.
15.46 Deben mantenerse registros de todas las
validaciones, calibraciones, mantenimiento, limpieza u operaciones de
reparación de los equipos principales y críticos, incluyendo fechas y la
identidad de las personas que llevaron a cabo estas operaciones.
15.47 El uso de equipos principales y críticos y las áreas
donde los productos han sido procesados debe registrarse apropiadamente en
orden cronológico.
15.48 Debe haber procedimientos operativos normatizados
asignando la responsabilidad de la limpieza y sanitización y describiendo con
suficiente detalle los programas de limpieza, métodos, equipos y materiales a
utilizarse e instalaciones y equipos a limpiar. Dichos procedimientos escritos
deben seguirse y registrarse.
16. BUENAS PRACTICAS EN PRODUCCION
16.1 Principio. Las operaciones de producción deben seguir
procedimientos claramente definidos de acuerdo con las autorizaciones de
fabricación y comercialización, con el objetivo de obtener productos de la
calidad requerida.
Generalidades
16.2 Toda manipulación de materiales y productos, tales
como recepción y limpieza, cuarentena, muestreo, almacenamiento, rotulado,
dispensación, procesamiento, acondicionado y distribución deben ser realizadas
de acuerdo a procedimientos o instrucciones escritas y registradas.
16.3 Cualquier desvío de las instrucciones o
procedimientos deben ser evitados. En caso de ocurrir, debe contarse con un
procedimiento operativo normatizado y la autorización debe ser aprobada por
escrito por una persona designada, con el compromiso del departamento de
control de calidad, según el caso.
16.4 Los controles en los rendimientos y las
reconciliaciones en las cantidades deben ser llevadas a cabo como una necesidad
de asegurar que no hay discrepancias por fuera de los límites aceptables.
16.5 Las operaciones sobre diferentes productos no deben
ser llevadas a cabo simultáneamente o consecutivamente en la misma sala o área.
16.6 Durante todo el tiempo de la elaboración, todos los
materiales, contenedores de graneles, principales partes del equipamiento,
salas y líneas de acondicionamiento a ser usadas deben estar rotuladas o
identificadas con indicación del producto o material a ser procesado, su
potencia /concentración, el número de lote, nombre y número de lote del
producto procesado previamente. Esta indicación debe también mencionar la etapa
de producción.
16.7 El acceso a los locales de producción debe estar
restringido a personal autorizado.
16.8 Normalmente, productos no medicinales no deberían ser
producidos en áreas o con equipos destinados para la producción de productos
farmacéuticos, salvo expresa autorización de la Autoridad Sanitaria.
16.9 Los controles en proceso son usualmente realizados en
el área de producción. La realización de tales controles en proceso no deben
tener ningún efecto negativo sobre la calidad del producto u otros productos
(ej. contaminación cruzada o mezcla).
Prevención de contaminación cruzada y bacteriana durante
la producción
16.10 Cuando son usados materiales y productos secos en la
producción, deben ser tomadas precauciones especiales para prevenir la
generación y diseminación de polvo. El suministro debe ser hecho por un control
de aire apropiado (ej. provisión y extracción de aire de calidad conveniente).
16.11 La contaminación de una materia prima o de un
producto por otra materia prima o producto debe ser evitada. El riesgo de una
contaminación surge de la liberación no controlada de polvo, gases, partículas,
vapores, sprays u organismos de los materiales y productos en proceso, de los
residuos en los equipos, de la entrada de insectos y de la ropa, piel, etc. del
operador. La trascendencia de los riesgos varía con el tipo de contaminante y
del producto que se contamina. Entre los contaminantes más peligrosos están los
materiales altamente sensibilizantes, preparaciones biológicas tales como organismos
vivos, ciertas hormonas, sustancias citostáticas y cualquier otro material
altamente activo. Los productos en los que la contaminación es más
significativa son aquellos que se administran por vía parenteral o se aplican
sobre heridas abiertas y los que se dan en dosis elevadas y/o por tiempo
prolongado.
16.12 La contaminación debe ser evitada tomando medidas
técnicas y de organización apropiadas, por ejemplo:
(a) llevando a cabo la producción en áreas dedicadas;
(b) produciendo en campaña (separadas en el tiempo)
seguidas de una adecuada limpieza de acuerdo con el procedimiento de limpieza
validado;
(c) proporcionando diseños adecuados de esclusas de aire,
diferenciales de presión y sistemas de suministro y extracción de aire;
(d) minimizando el riesgo de contaminación causada por la
recirculación o re-ingreso de aire no tratado o insuficientemente tratado;
(e) vistiendo ropa protectora donde los productos o
materiales son manipulados;
(f) usando procedimientos de limpieza y decontaminación
validados;
(g) usando en producción un "sistema cerrado";
(h) controlando los residuos;
(i) usando rótulos de condición de estado de limpieza en
los equipos.
16.13 Las medidas para la prevención de la contaminación y
su efectividad deben ser controladas periódicamente de acuerdo al procedimiento
operativo normatizado correspondiente.
16.14 Las áreas de producción donde se procesan productos
susceptibles deberían tener un monitoreo ambiental periódico (ej. monitoreo
microbiológico y de partículas).
Operaciones de procesamiento
16.15 Antes de que cualquier operación de procesamiento
comience, se debe asegurar que el área de trabajo y el equipamiento están
limpios y libres de cualquier materia prima, producto, residuo de producto,
rótulos o documentos que no sean requeridos para la operación actual.
16.16 Tanto los controles en proceso necesarios como los
controles ambientales deben ser llevados a cabo y registrados.
16.17 Se debe instituir la documentación de fallas de
equipos y de sus servicios (ej. agua, gas). Los equipos defectuosos deben ser
retirados de uso hasta que el defecto haya sido rectificado. Después del uso,
los equipos de producción deben ser limpiados sin demora de acuerdo a
procedimientos operativos normatizados y ser guardados limpios y secos en un
área separada o de manera que se prevenga la contaminación.
16.18 Los límites de tiempo para el guardado del equipo
después de la limpieza y antes del uso deben estar justificados.
16.19 Los contenedores a ser llenados deben estar limpios
antes del llenado. Debe prestarse atención para evitar y retirar cualquier tipo
de contaminante tales como fragmentos de vidrio y partículas metálicas.
16.20 Cualquier desvío significativo del rendimiento
esperado debe ser registrado e investigado.
16.21 Se deben llevar a cabo controles para asegurar que
las tuberías y otras piezas del equipo usado para el transporte de productos de
un área a otra están conectados de forma correcta.
16.22 Las tuberías utilizadas para el transporte de agua
destilada o deionizada y/o de otra calidad de agua deben ser sanitizadas y
guardadas, cuando corresponda, de acuerdo a procedimientos operativos
normatizados que detallen los límites de acción para la contaminación
microbiológica y las medidas a tomar.
16.23 Los equipos de control e instrumentos de medición,
pesaje y registro deben ser mantenidos y calibrados en intervalos
preestablecidos y con registros de los mismos. Para asegurar un funcionamiento
satisfactorio, los instrumentos deben ser controlados diariamente o previo a su
uso realizando ensayos analíticos. Los datos de calibración y mantenimiento y
la fecha de re–calibración deben estar claramente indicados en un rótulo
adherido al instrumento.
16.24 Las operaciones de reparación y mantenimiento no
deben presentar ningún peligro para la calidad de los productos.
Operaciones de acondicionamiento
16.25 Cuando se establece el programa de operaciones de
acondicionamiento, se debe prestar particular atención para minimizar los
riesgos de contaminación, mezclas o sustituciones. No se deben acondicionar
diferentes productos en proximidades cercanas a menos que haya una separación
física que lo asegure.
16.26 Antes de que las operaciones de acondicionamiento
comiencen, se debe asegurar que el área de trabajo, las líneas de acondicionamiento,
las impresoras y otros equipos estén limpios y libres de otros productos,
materiales o documentos usados anteriormente y que no son requeridos para la
operación actual. La liberación de la línea debe ser realizada de acuerdo a un
procedimiento operativo normatizado y a una lista de verificación, con
registros.
16.27 El nombre, forma farmacéutica, concentración y
número de lote del producto que se está manejando debe ser exhibido en cada
estación o línea de acondicionamiento.
16.28 El llenado y cerrado debe realizarse sobre envases
ya codificados (rotulado), por lo menos con el nombre del producto y su número
de lote, a menos que ambas operaciones (llenado/cerrado y rotulado) se realicen
en línea (proceso continuo).
16.28.1 En caso de no contar con envases ya codificados
(rotulados), la identificación debe realizarse en forma inmediata después del
llenado y cerrado.
16.29 La realización correcta de cualquier impresión (ej.
código de números o fecha de vencimiento) debe estar controlada y registrada. Se
debe prestar atención a la impresión manual, la que debe ser re-controlada a
intervalos regulares.
16.30 Se debe tener especial cuidado cuando se usan
rótulos cortados, y se realizan las operaciones de acondicionamiento
manualmente. Se prefieren los rollos de rótulos a los rótulos cortados para
ayudar a impedir las mezclas o confusiones. La verificación en línea de todas
los rótulos por medio de sistemas electrónicos automáticos puede ser de ayuda
para la prevención de mezclas o confusiones, pero se debe controlar para
asegurar que las lectoras electrónicas de códigos, contadores de rótulos o
cualquier aparato similar están funcionando correctamente. Cuando los rótulos
son pegados en forma manual, los controles en proceso se deben realizar mas
frecuentemente.
16.31 La información impresa y realzada en relieve de los
materiales de acondicionamiento debe ser clara y resistente a la decoloración o
borrado.
16.32 El control regular en línea del producto durante el
acondicionamiento debe incluir al menos controles de:
(a) apariencia general de los envases;
(b) si los envases están completos;
(c) si son usados los productos y materiales de
acondicionamiento correctos;
(d) el funcionamiento correcto de los monitores en línea.
(e) si la impresión de lote y vencimiento es correcta.
Las muestras que se retiren de la línea de
acondicionamiento no deben ser reingresadas a ella, sin autorización previa.
16.33 Los productos que han sido involucrados en un evento
inusual durante el acondicionamiento deben ser re-introducidos dentro del
proceso sólo después de una inspección especial, investigación y aprobación por
personal autorizado. Se debe guardar un registro detallado de esta operación.
16.34 Cualquier discrepancia significativa o inusual
observada durante la conciliación de la cantidad de producto en granel,
material de acondicionamiento impreso y el número de unidades producidas debe
ser investigada, satisfactoriamente explicada y registrada antes de la
liberación.
16.35 Al completar la operación de acondicionamiento,
cualquier material de acondicionamiento codificado con el lote debe ser
destruido y la destrucción registrada. Se debe seguir un procedimiento
operativo normatizado con los controles exigidos a ser realizados antes de re–
ingresar materiales no usados, es decir si los materiales impresos no
codificados son regresados al stock.
Reprocesamiento y recuperación de lotes
16.36 El procedimiento general de reprocesamiento debe
estar controlado por un Procedimiento Operativo Normatizado de desvíos.
El reprocesamiento de productos rechazados debe ser una
medida excepcional. El reprocesamiento sólo puede ser permitido si se demuestra
que la calidad especificada del producto final no resulta afectada y debe ser
realizado según un procedimiento definido y autorizado tras una evaluación de
los riesgos implicados. Debe conservarse un registro de la operación de
reprocesamiento.
16.37 La recuperación se debe realizar en base a un
procedimiento definido tras una evaluación de los riesgos implicados,
incluyendo cualquier posible efecto sobre la caducidad. La introducción total o
parcial de lotes anteriores, conformes a la calidad requerida, en un lote del
mismo producto en una fase determinada de la fabricación debe ser autorizada
previamente. Esta recuperación debe ser registrada.
16.38 El departamento de control de calidad debe
considerar la necesidad de realizar pruebas suplementarias de cualquier
producto terminado que se haya reprocesado o al que se le haya incorporado un
producto recuperado.
17. BUENAS PRACTICAS EN CONTROL DE CALIDAD
17.1 En control de calidad se encuentran involucrados el
muestreo, especificaciones y ensayos, como así también los procedimientos de la
organización, documentación y autorización que aseguren que los ensayos necesarios
y relevantes son llevados a cabo y que no se autorice el uso de materiales ni
la expedición de productos para su distribución o venta, sin que se halla
establecido que su calidad es satisfactoria. Control de calidad no se confina a
operaciones de laboratorio, debe estar involucrada en todas las decisiones
concernientes a la calidad del producto.
17.2 La independencia entre control de calidad y
producción se considera fundamental.
17.3 Cada elaborador (titular de la autorización de
elaboración) debe tener una función de control de calidad. La función de
control de calidad debe ser independiente de otros departamentos y estar bajo
la autoridad de una persona con cualidades y experiencia apropiada, quien tenga
uno o varios laboratorios de control a su disposición. Los recursos adecuados
deben estar disponibles para asegurar que todas las medidas de control de
calidad son efectivas y son llevadas a cabo en forma confiable. Los
requerimientos básicos para control de calidad son los que siguen:
(a) instalaciones adecuadas, personal entrenado y
procedimientos aprobados deben estar disponibles para muestro, inspecciones y
control de materias primas, material de acondicionamiento y producto
intermedio, granel y terminado, así como para el monitoreo de las condiciones
ambientales para el cumplimiento de las BPF;
(b) las muestras de materia prima, material de
acondicionamiento, producto intermedio, a granel y terminado deben ser tomadas
por métodos y personal aprobado por el departamento de control de calidad, según
PON;
(c) se debe realizar calificación de equipos e
instrumentos y validación de metodologías;
(d) se deben realizar registros (manualmente y/o por
instrumentos), demostrando que todos los procedimientos de muestreo, inspección
y control se han llevado a cabo y que cualquier desvío ha sido completamente
registrado e investigado;
(e) los productos terminados deben contener los
ingredientes preestablecidos en la composición cuali-cuantitativa del producto
autorizados para su comercialización por la Autoridad Sanitaria Nacional; los ingredientes deben ser de la pureza requerida, estar en el
contenedor adecuado y correctamente etiquetado;
(f) se deben registrar los resultados de inspecciones y
controles de materiales y producto intermedio, granel y terminado según
especificaciones; la evaluación del producto debe incluir la revisión y
evaluación de la documentación de producción, como así también de los desvíos
de los procedimientos especificados;
(g) ningún lote de producto es liberado para la venta o suministro
sin la certificación por la/s persona/s autorizada/s de que esta de acuerdo con
los requerimientos de la autorización de comercialización;
(h) se deben conservar suficientes muestras de materias
primas y productos terminados para permitir realizar por triplicado los
controles que aseguran la calidad del producto; los productos conservados se
deben guardar en su envase final a menos que el envase sea excepcionalmente
grande, por un período no inferior a un año posterior a su fecha de vencimiento.
17.4 Control de calidad tiene entre otras tareas, las de
establecer, validar e implementar todos los procedimientos de control de
calidad; evaluar, mantener y almacenar los sustancias y materiales de
referencia, asegurar el correcto etiquetado de los contenedores de los
materiales y productos, asegurar el monitoreo de la estabilidad de los
ingredientes farmacológicamente activos y de los productos; participar en la
investigación de las quejas relacionadas con la calidad del producto y
participar en el monitoreo ambiental. Todas estas operaciones deben ser
llevadas a cabo de acuerdo a procedimientos operativos normatizados y
registrados.
17.5 La evaluación de los productos terminados debe
abarcar todos los factores relevantes, incluyendo las condiciones de producción,
los resultados de los controles en proceso, la documentación de la elaboración
(incluyendo el acondicionamiento), cumpliendo con las especificaciones para
producto terminado, y un examen del envase final.
17.6 El personal de control de calidad debe tener acceso a
las áreas de producción para un muestreo e investigación apropiada.
Control de materias primas, productos intermedio, a granel
y terminado
17.7 Todos los ensayos deben seguir las instrucciones
establecidas en los procedimientos operativos normatizados para cada material o
producto. Los resultados deben ser controlados por el supervisor antes de que
el material o producto sea liberado o rechazado.
17.8 Las muestras deben ser representativas de los lotes
del material del cual fueron tomadas de acuerdo con el procedimiento operativo
normatizado aprobado.
17.9 El muestreo debe ser llevado a cabo de forma tal de
evitar la contaminación u otros factores que puedan afectar la calidad. Los
contenedores que han sido muestreados deben estar identificados de conformidad
a un procedimiento operativo normatizado y vueltos a cerrar correctamente
después del muestreo.
17.10 Durante la operación de muestreo se debe tener la
precaución de resguardar el material a ser muestreado de la contaminación y
mezcla. Todo el equipo de muestreo que entra en contacto con el material debe
estar limpio. En caso de materiales particularmente tóxicos, peligrosos,
sensibilizantes, etc. se deben seguir precauciones especiales preestablecidas
en un procedimiento operativo normatizado.
17.11 El equipo de muestreo debe estar limpio, y si es
necesario, esterilizado antes y después de cada uso y almacenados separadamente
de otros equipos de laboratorio.
17.12 Cada contenedor de muestra debe poseer una rotulo
que indique como mínimo:
(a) el nombre del material muestreado;
(b) el número de lote;
(c) el número del contenedor del cual se ha tomado la
muestra;
(d) el número de la muestra;
(e) la firma de la persona que ha tomado la muestra;
(f) la fecha de muestreo.
17.13 Los resultados fuera de especificación obtenidos
durante el control de materiales o productos deben ser investigados de acuerdo
con procedimientos operativos normatizados aprobados. Se deben mantener
registros.
Requisitos de los controles
Materias primas y material de acondicionamiento
17.14 Antes de la liberación de una materia prima o
material de acondicionamiento para su uso, el encargado de control de calidad
debe asegurar que los materiales han sido controlados conforme con las
especificaciones para identificación, potencia, pureza y otros parámetros de
calidad.
17.15 Se debe realizar un ensayo de identificación sobre
una muestra de cada contenedor de materia prima (ver sección 14.14).
17.16 Cada lote de material de acondicionamiento impreso
debe ser examinado después de recibido.
17.17 Los certificados de análisis deben contener al menos
la siguiente información:
(a) el nombre del material analizado;
(b) el número de lote del material analizado;
(c) las especificaciones y métodos usados;
(d) los resultados obtenidos;
(e) conclusión;
(f) la fecha de comienzo y de fin del análisis.
Controles en proceso
17.18 Los registros de los controles en proceso deben ser
mantenidos y formar parte de los registros del lote.
Productos terminados
17.19 Antes de la liberación de cada lote de producto,
debe haber una aprobación por escrito por parte del responsable de control de
calidad que el producto cumple con las especificaciones preestablecidas.
17.20 Los productos que no cumplan con las especificaciones
o cualquier otro criterio de calidad relevante deben ser rechazados.
Revisión del registro de lote
17.21 Los registros de producción y control de calidad
deben ser revisados como parte del proceso de aprobación de la liberación del
lote. Cualquier divergencia con las especificaciones debe ser profundamente
investigada. La investigación debe, si es necesario, extenderse a otros lotes
del mismo producto y otros productos que pudieran estar asociados con la
discrepancia. Se debe llevar un registro escrito de la investigación e incluir
la conclusión y la acción a seguir.
17.22 Las muestras conservadas de cada lote de producto
terminado deben ser mantenidas al menos por un año después de la fecha de
vencimiento. Los productos terminados deben ser guardados en su envase final y
almacenados bajo las condiciones recomendadas. Si envases excepcionalmente
grandes son producidos, se podrían almacenar pequeñas muestras en contenedores
apropiados. Las muestras de materias primas activas deben ser conservadas por
al menos un año después de la fecha de vencimiento del correspondiente producto
terminado. Otras materias primas (a parte de solventes, gases y agua) deberían
conservarse por un mínimo de dos años si su estabilidad lo permite. Las
muestras conservadas de materiales y productos deben ser de un tamaño
suficiente que permita al menos tres exámenes completos.
Estudios de estabilidad
17.23 Control de calidad debe evaluar la calidad y
estabilidad de los productos farmacéuticos terminados y, cuando sea necesario,
de las materias primas y productos intermedios.
17.24 Control de calidad debe proponer las fechas de
vencimiento y las especificaciones de vida útil en base a los estudios de
estabilidad relacionados a las condiciones de almacenamiento. Lo propuesto
queda sujeto a consideración de la Autoridad Sanitaria Nacional.
17.25 Se debe desarrollar e implementar un programa
escrito para llevar a cabo la determinación de estabilidad que incluya como
mínimo los siguientes elementos:
(a) una descripción completa de la droga involucrada en el
estudio;
(b) la batería completa de métodos y paramentos a
analizar, describiendo todos los análisis para potencia, pureza,
características físicas y evidencia documentada de que los mismos indican
estabilidad;
(c) la inclusión de un número suficiente de lotes;
(d) el esquema de análisis para cada droga;
(e) abastecimiento para condiciones especiales de
almacenamiento;
(f) abastecimiento para conservar muestras adecuadas;
(g) resumen de todos los datos generados, incluyendo la evaluación
y las conclusiones del estudio.
17.26 La estabilidad debe ser determinada antes de la
comercialización y seguir cualquier cambio significativo en procesos, equipos,
material de empaque, etc.
18. PRODUCTOS FARMACEUTICOS ESTERILES
Introducción
18.1 La elaboración de medicamentos estériles está sujeta
a requisitos especiales para minimizar los riesgos de contaminación microbiana,
de partículas y de sustancias pirogénicas.
Generalidades
18.2 La elaboración de preparaciones estériles debe
llevarse a cabo en áreas limpias; el ingreso a las cuales debe efectuarse a
través de esclusas, tanto para el personal como para los materiales y equipos.
Las áreas limpias deben mantenerse de conformidad con normas apropiadas de
limpieza, y se debe suministrar a las mismas solamente aire que ha pasado a
través de filtros de eficiencia comprobada.
18.3 Las diversas operaciones de preparación de los
componentes (tales como recipientes y cierres), preparación de productos,
llenado y esterilización deben llevarse a cabo en áreas separadas, debiendo ser
áreas limpias. Estas áreas son clasificadas en cuatro grados (ver tabla 1).
18.4 Las áreas limpias para la elaboración de productos
estériles están clasificadas de acuerdo a las características del ambiente
requeridas. Cada operación de elaboración requiere de un determinado grado de
limpieza para minimizar el riesgo de contaminación por partículas o
microbiológico de los productos o materiales que están siendo manipulados.
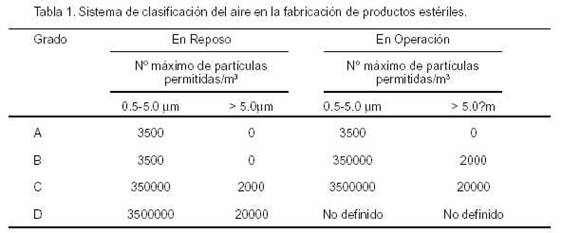
Para reunir las condiciones de "en operación",
estas áreas deben estar diseñadas para alcanzar ciertos niveles especificados
de aire limpio en el estado de "en reposo". Este último estado es la
condición donde la instalación está completa, y el equipamiento de producción
ha sido instalado y está operando, pero no está presente el personal. El estado
de "en operación" es la condición donde la instalación está funcionando
en el modo operativo definido y el número de personal especificado está
presente.
• Grado A: Es el área para operaciones de alto riesgo,
ejemplo: llenado y preparación asépticas. Normalmente tales condiciones son
provistas por una estación de trabajo de flujo laminar. Los sistemas de
corriente de aire laminar deben suministrar una velocidad de aire homogénea de
aproximadamente 0,45 m/s +/- 20% (valor orientativo) en la posición de trabajo.
• Grado B: Es el área que rodea a la de grado A en llenado
y preparaciones asépticas.
• Grado C y D: Son las áreas limpias para llevar a cabo
los pasos menos críticos de la elaboración de productos estériles.
• Para alcanzar los grados de aire B, C y D, el número de
cambios de aire debe ser apropiado para el tamaño del área y del equipo y
personal presente en ella. Al menos 20 cambios de aire por hora son requeridos
para áreas con un buen patrón de corriente de aire y filtros de aire de alta
eficacia (HEPA).
18.5 Las condiciones de partículas indicadas en la tabla 1
para la situación "en reposo", deben recuperarse en ausencia de
personal tras un breve período de "limpieza" de 15-20 minutos (valor
orientativo), una vez finalizadas las operaciones. Las condiciones de
partículas para el grado A "en operación" que figuran en la tabla 1
deben mantenerse en la zona inmediata que rodea al producto, siempre que el
producto o envase abierto esté expuesto al entorno. Se acepta que no siempre
puede demostrarse la conformidad con las normas de partículas en el punto de
llenado, debido a la producción de partículas procedentes del producto.
18.6 Se deben monitorear/supervisar las diferentes áreas
limpias durante la operación, a fin de controlar el nivel de partículas de los
distintos grados.
18.7 Se deben establecer límites de alerta y de acción
apropiados para los resultados del monitoreo de partículas y microbiológicos.
Si éstos se exceden se deben tomar las acciones correctivas apropiadas según
procedimientos operativo normatizados.
18.8 Los grados de las áreas como se han especificado,
deben ser elegidos por el elaborador en base a la naturaleza de las operaciones
del proceso y a las validaciones (ej. media fills). La determinación de un área
y límite de tiempo apropiados para el proceso se debe basar en la contaminación
microbiológica y recuento de partículas encontrado.
18.9 Durante las operaciones, las áreas limpias deben
controlarse a intervalos preestablecidos, mediante el recuento microbiano del
aire y de las superficies como para asegurar que el ambiente esté dentro de las
especificaciones. Se deben usar métodos tales como placas de exposición, aire
forzado, muestreo de superficies (por ej. hisopos y placas de contacto). Las
zonas no deben contaminarse por los métodos de muestreo utilizados en la
operación. Deben tenerse en cuenta los resultados del control ambiental en la
evaluación de los lotes para su posterior autorización. Tanto las superficies
como el personal deben ser monitoreados después de operaciones críticas. Se
debe controlar también regularmente la calidad del aire con respecto al
contenido de partículas. Es conveniente efectuar controles adicionales, aun
cuando no se efectúen operaciones de producción, como por ejemplo después de la
validación de los sistemas, de la limpieza y de la fumigación.
18.10 Los niveles de detección de contaminación
microbiológica deben ser establecidos como límites de alerta y de acción para
monitorear la calidad del aire en las áreas. En la tabla 2 son dados los
límites para el monitoreo microbiológico de áreas limpias en operación
expresados en unidades formadoras de colonia (UFC). Los métodos de muestreo y
los valores que incluye la tabla no representan especificaciones, son
recomendaciones.
Tabla 2. Límites para el monitoreo de contaminación
microbiología.
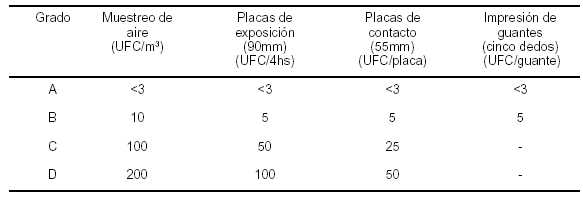
Elaboración de productos estériles
18.11 Cada operación de fabricación requiere un nivel
apropiado de limpieza del aire, para reducir al mínimo los riesgos de contaminación
microbiana o por partículas del producto o de los materiales que se están
manipulando.
18.12 Las operaciones de elaboración se desarrollan según:
(a) Aquellas donde el producto es esterilizado en forma
terminal;
(b) Aquellas en las que el producto se elabora de forma
aséptica en alguna etapa (ej. Luego de filtración esterilizante) o en todas sus
etapas (ej. A partir de materias primas estériles);
(c) Aquellas en que se utiliza tecnología de aislador en
alguna/s o en todas sus etapas;
(d) Aquellas en que se utiliza tecnología de
soplado/llenado/sellado.
Productos esterilizados en forma terminal
18.13 La preparación de componentes y de la mayoría de los
productos debe realizarse, al menos, en un ambiente grado D, con el objeto de
obtener recuentos microbianos y de partículas bajos, siempre y cuando se
sometan a filtración antes de su llenado y posteriormente sean esterilizados en
forma terminal.
18.14 Cuando el producto tenga un riesgo elevado o inusual
de contaminación microbianana por ejemplo, porque el producto favorezca
activamente el crecimiento microbiano o deba pasar mucho tiempo antes de la
esterilización o se elabore en recipientes no cerrados, la preparación debe
efectuarse en un entorno de grado C, siempre y cuando se sometan a filtración
antes de su llenado y posteriormente sean esterilizados en forma terminal.
18.15 El llenado de productos parenterales esterilizados
al final del proceso debe realizarse en un ambiente grado A con un entorno, al
menos, de grado C.
18.16 La preparación y el llenado de ungüentos, pomadas,
cremas, suspensiones y emulsiones, deben hacerse en un ambiente grado C antes
de la esterilización terminal.
18.17 Cuando para los productos mencionados en el punto
anterior, exista un riesgo inusual de contaminación por el entorno, por
ejemplo, debido a que la operación de llenado sea lenta o los recipientes
tengan cuello ancho o estén expuestos necesariamente durante más de unos
segundos antes de su cierre, el llenado debe hacerse en una zona de grado A con
un entorno al menos de grado C.
Preparación aséptica
18.18 Una vez lavados, los componentes deben manipularse
en una zona de grado D. La manipulación de componentes y materiales de partida
estériles, salvo que se sometan a esterilización o filtración a través de un
filtro que retenga los microorganismos en una fase posterior del proceso, debe
realizarse en grado A con entorno grado B.
18.19 La preparación de soluciones que deban esterilizarse
por filtración durante el proceso debe hacerse en grado C. Si no se filtran, la
preparación de materiales y productos debe hacerse en grado A con entorno de
grado B.
18.20 La manipulación y el llenado de productos preparados
asépticamente deben hacerse en grado A con entorno de grado B.
18.21 Luego de la filtración esterilizante, el producto
debe manipularse y llenarse en recipientes bajo condiciones estériles en un
área de grado A, con ambiente de grado B.
18.22 La transferencia de recipientes parcialmente
cerrados, como los utilizados en la liofilización, antes de completar su
cerrado, debe hacerse en grado A con entorno de grado B, o bien en bandejas de
transporte cerradas en grado A y transportadas en zona grado B.
18.23 La preparación y llenado de ungüentos, pomadas,
cremas, suspensiones y emulsiones estériles deben hacerse en grado A con
entorno de grado B, cuando el producto esté expuesto y no se filtre
posteriormente.
Tecnología de aislador
18.24 La utilización de la tecnología de aislador para
reducir las intervenciones humanas en las zonas de elaboración puede producir
un descenso significativo del riesgo de contaminación microbiológica procedente
del entorno en los productos de elaboración aséptica. El aislador y su entorno
deben diseñarse de forma que pueda alcanzarse la calidad de aire requerida en
las zonas respectivas. El equipo de transferencia puede variar entre diseños de
una puerta simple o doble, hasta sistemas totalmente herméticos que incorporan
mecanismos de esterilización.
18.25 La entrada y salida de materiales de la unidad
constituye una de las mayores fuentes posibles de contaminación. El área del
interior del aislador es el lugar donde se hacen las manipulaciones de riesgo
elevado. La clasificación de aire requerida para el entorno depende del diseño
del aislador y de su aplicación. Debe controlarse y debe ser al menos de grado
D.
18.26 La transferencia de recipientes parcialmente
cerrados debe hacerse en un área de grado A con entorno de grado B, antes de
completar su cerrado.
18.27 Los aisladores deben utilizarse sólo después de una
validación adecuada. Esta validación debe tener en cuenta todos los factores
críticos de la tecnología de los aisladores, por ejemplo la calidad de aire del
interior y exterior.
18.28 El monitoreo debe realizarse de forma sistemática y
según procedimientos operativos normatizados. Debe incluir pruebas frecuentes
de ausencia de fugas del aislador y del sistema de guantemanga.
Tecnología de soplado/llenado/sellado
18.29 Las unidades de soplado/llenado/sellado son equipos
diseñados específicamente para que en una operación continua se formen los
recipientes a partir de un granulado termoplástico, se llenan y se sellen, en
una sola máquina automática. Este equipo debe estar provisto de un chorro
eficaz de aire de grado A. En el caso de preparaciones asépticas, el equipo
debe instalarse en un entorno al menos de grado C, utilizando vestimenta de
grado A/B. El equipo de soplado/llenado/sellado utilizado para la elaboración
de productos esterilizados al final del proceso debe instalarse en un entorno
al menos de grado D.
18.30 Con esta tecnología, debe prestarse especial
atención a los siguientes puntos, entre otros:
(a) diseño y calificación del equipo;
(b) validación de la limpieza y de la esterilización;
(c) clasificación del entorno de área limpia donde se
encuentre el equipo;
(d) formación y vestimenta de los operadores e
intervenciones en la zona crítica del equipo, incluido el montaje aséptico
antes del comienzo de la operación de llenado.
Proceso
18.31 Durante todas las etapas del proceso deben adoptarse
precauciones para minimizar la contaminación, incluso durante las etapas
anteriores a la esterilización.
18.32 No deben fabricarse preparaciones que contengan
microorganismos vivos en áreas usadas para el procesamiento de otros productos
farmacéuticos, ni tampoco efectuarse el llenado de recipien- tes con dichas
preparaciones; sin embargo, puede efectuarse el llenado de recipientes con
vacunas de organismos inactivados o de extractos bacterianos en el mismo
recinto que otros productos farmacéuticos estériles, siempre que la
inactivación haya sido validada y se hayan efectuado procedimientos validados
de limpieza.
18.33 La validación del proceso aséptico debe incluir la
simulación del proceso utilizando un medio nutritivo (media-fills), esta
simulación se debe realizar como una validación inicial con tres ensayos de
simulación satisfactorios consecutivos. La simulación del proceso debe
repetirse a intervalos definidos y después de cualquier modificación
significativa del equipo y proceso. La selección del medio nutritivo utilizado
debe estar basada en la forma farmacéutica del producto y en la selectividad,
claridad, concentración y adecuación del medio para la esterilización. La
prueba de simulación del proceso debe imitar, lo más exactamente posible, el
proceso de fabricación aséptica normal e incluir todas las fases críticas
posteriores de la fabricación. El número de envases utilizados para llenado con
el medio de cultivo debe ser suficiente para que la evaluación sea válida. La
tasa de contaminación debe ser inferior al 0,1% con un nivel de confianza del
95%. El elaborador debe establecer límites de alerta y de acción. Cualquier
contaminación debe ser investigada.
18.34 Debe procurarse que las validaciones no pongan en
peligro el proceso de elaboración.
18.35 Deben controlarse regularmente las fuentes de
provisión de agua, los equipos de tratamiento de agua y el agua tratada, para
verificar si existen sustancias químicas, contaminación biológica, o contaminación
con endotoxinas, con el fin de asegurar, antes de usarla, que el agua cumple
con las especificaciones correspondientes al uso que se le quiere dar. Deben
mantenerse registros de los resultados obtenidos y de las medidas adoptadas.
18.36 Las actividades efectuadas en áreas limpias
clasificadas deben reducirse al mínimo, especialmente cuando se están
efectuando operaciones asépticas y el movimiento de personal debe ser metódico
y controlado, con el fin de evitar el excesivo desprendimiento de partículas y
microorganismos. Debido a la naturaleza de la vestimenta empleada, la
temperatura y la humedad del ambiente no deben ser tan altas que causen
incomodidad.
18.37 La contaminación microbiológica de las materias
primas debe ser mínima. Las especificaciones deben incluir requisitos de
calidad microbiológica.
18.38 La presencia de recipientes y materiales que pueden
desprender fibras debe reducirse al mínimo en las áreas estériles y evitarse
completamente cuando se está efectuando un trabajo aséptico.
18.39 Después del proceso final de esterilización, el
manejo de los componentes, recipientes de productos a granel y equipos debe
efectuarse de tal forma que no se contaminen nuevamente. Debe identificarse
inequívocamente la etapa del procesado de componentes, recipientes de productos
a granel y equipos.
18.40 El intervalo entre el lavado y el secado y la
esterilización de los componentes, recipientes de productos a granel y otros
equipos, como también el intervalo entre la esterilización y el uso, deben ser
lo más breves posibles y estar sometidos a un límite de tiempo acorde con las
condiciones validadas de almacenamiento.
18.41 El tiempo transcurrido entre el inicio de la
preparación de una solución y su esterilización o filtración a través de
filtros de retención de bacterias debe ser lo más breve posible y debe ser
validado.
18.42 Todo gas utilizado para purgar una solución o un
envase debe pasarse a través de un filtro esterilizante de comprobada
eficiencia.
18.43 La carga biológica de los productos debe ser mínima
antes de la esterilización. Debe establecerse el límite funcional al que puede
llegar la contaminación inmediatamente antes de la esterilización, el cual debe
estar relacionado con la eficiencia del método a ser empleado y con el riesgo
de sustancias pirogénicas. Todas las soluciones, especialmente las parenterales
de gran volumen, deben pasar por un filtro de retención microbiana, de ser
posible inmediatamente antes del proceso de llenado. Cuando se trata de
soluciones acuosas en recipientes cerrados herméticamente, los orificios
compensadores de presión deben estar protegidos con filtros de venteo
esterilizadores hidrófobos.
18.44 Todos los componentes, recipientes de productos a
granel, equipos y cualquier otro artículo que sea necesario en las áreas
limpias clasificadas donde se efectúan trabajos asépticos se deben esterilizar
y, de ser posible, introducir a dichas áreas a través de equipos de
esterilización de doble puerta embutidos en la pared y que descarguen
directamente al área limpia clasificada. En algunas circunstancias podrían ser
aceptables otros procedimientos que dan los mismos resultados en lo que
respecta a impedir la contaminación.
18.45 Debe validarse la eficacia de cualquier sistema de
procesamiento nuevo y esa validación debe repetirse a intervalos programados, y
cuando se haga un cambio importante en el proceso o en los equipos utilizados.
Esterilización
18.46 Se puede efectuar la esterilización por medio del
calor húmedo o seco, del óxido de etileno (u otro agente esterilizante gaseoso
apropiado), por filtración y el subsiguiente llenado aséptico de los
recipientes finales estériles o por irradiación con radiación ionizante (pero
no con radiación ultravioleta, a menos que este procedimiento haya sido
totalmente validado). Cada método tiene sus aplicaciones y limitaciones
particulares. De ser posible y conveniente, el método de elección debe ser la
esterilización térmica.
18.47 Todos los procedimientos de esterilización deben ser
validados. Se debe prestar especial atención cuando el método de esterilización
adoptado se emplea con una preparación que no sea una simple solución acuosa o
aceitosa. En todos los casos, el método de esterilización debe estar de acuerdo
con la respectiva autorización de comercialización.
18.48 La contaminación microbiológica de las materias
primas debe ser mínima y debe ser monitoreada antes de la esterilización.
18.49 Antes de aprobar un método de esterilización, debe
demostrarse que es adecuado para el producto en cuestión y que es eficaz para
alcanzar los niveles de esterilización deseados en todas las partes de cada
tipo de carga a ser procesada. La validez del proceso debe verificarse a
intervalos preestablecidos, o anualmente como mínimo, y siempre que se han
introducido modificaciones importantes en los equipos. Asimismo, deben
mantenerse registros de los resultados obtenidos.
18.50 Los indicadores biológicos deben ser considerados
solamente como factores adicionales para el control de la esterilización. En
caso de que se utilicen, deben tomarse precauciones estrictas para evitar que
sean causa de contaminación microbiana.
18.51 Se debe contar con un medio inequívoco de distinguir
los productos que han sido esterilizados de los que no lo han sido. Cada
canasta, bandeja, u otro tipo de transportador debe ser claramente rotulado con
el nombre del material, el número de lote y una indicación de si ha sido o no
esterilizado. Pueden usarse indicadores tales como cinta de autoclave, cuando
sea apropiado, para indicar si un lote ha sido sometido o no a un proceso de
esterilización, pero este sistema no proporciona una indicación confiable de
que un lote es, en realidad, estéril.
18.52 Debe haber registros de la esterilización de cada
ciclo de esterilización. Estos registros se aprobaran como parte del procedimiento
de aprobación del lote.
ESTERILIZACION TERMINAL
Esterilización térmica
18.53 Cada ciclo de esterilización térmica debe ser
registrado mediante equipos apropiados, y con la debida precisión, como por
ejemplo, en una tabla de tiempo/temperatura con una escala de tamaño adecuado.
La temperatura debe registrarse mediante una sonda colocada en el punto más
frío de la carga o de la cámara cargada, habiéndose determinado este punto
durante la validación; preferiblemente la temperatura debe ser verificada,
comparándola con otra temperatura tomada mediante otra sonda independiente
colocada en la misma posición. La mencionada tabla de tiempo/temperatura, o
bien una fotocopia de la misma, debe formar parte del registro del lote. Pueden
emplearse también indicadores químicos o biológicos, pero éstos no deben
reemplazar a los controles efectuados por medios físicos.
18.54 Se debe dejar transcurrir suficiente tiempo para que
toda la carga alcance la temperatura requerida antes de empezar a medir el
tiempo de esterilización. Para cada tipo de carga debe determinarse dicho
tiempo.
18.55 Luego de la etapa de alta temperatura de un ciclo de
esterilización térmica, se deben tomar precauciones para evitar que una carga
esterilizada se contamine durante el enfriamiento.
Esterilización con calor húmedo
18.56 La esterilización con calor húmedo es apropiada
solamente para materiales que pueden mojarse con agua y para soluciones
acuosas. Para controlar este proceso deben tenerse en cuenta tanto la
temperatura como la presión. Normalmente el instrumento que registra la
temperatura debe ser independiente del utilizado para el control y se debe
utilizar un indicador de temperatura también independiente, cuya lectura debe
compararse regularmente con el registrador de la tabla durante el período de
esterilización. Si se trata de esterilizadores que tienen un drenaje en el
fondo de la cámara, tal vez sea necesario registrar también la temperatura en
esta posición, durante todo el período de esterilización. Cuando forma parte
del ciclo una fase al vacío, entonces deben efectuarse controles regulares para
verificar si la cámara tiene pérdidas.
18.57 Los productos a ser esterilizados, siempre que no se
trate de recipientes herméticamente cerrados, deben envolverse en un material
que permita la eliminación del aire y la penetración de vapor, pero que impida
la recontaminación después de la esterilización. Todas las partes de la carga
deben estar en contacto con el agua o el vapor saturado a la temperatura
requerida y por el tiempo requerido.
18.58 Se debe asegurar que el vapor empleado en la
esterilización sea de la calidad adecuada y que no contenga aditivos en un
nivel tal que puedan ser causa de contaminación del producto o de los equipos.
Esterilización con calor seco
18.59 Cuando se emplea el proceso de esterilización con
calor seco, el aire debe circular dentro de la cámara, manteniéndose una
presión positiva para impedir la entrada de aire no estéril. El aire
suministrado debe ser pasado por un filtro que retenga microorganismos (ej.
filtro HEPA). Si el proceso de esterilización con calor seco tiene por objeto
también la eliminación de pirógenos, como parte de la validación deberán
efectuarse pruebas de desafío empleando endotoxinas.
Esterilización por radiación
18.60 La esterilización por radiación se usa
principalmente para la esterilización de materiales y productos sensibles al
calor. Debido a que muchos productos farmacéuticos y materiales de envasado son
sensibles a la radiación, se permite emplear este método cuando la ausencia de
efectos nocivos sobre el producto ha sido confirmada experimentalmente. La
radiación ultravioleta no es un método aceptable de esterilización terminal.
18.61 Si la esterilización por radiación se encarga a un
contratista independiente, el fabricante es responsable de asegurar que se
cumplan las normas de la Sección anterior, y que el proceso de esterilización
sea validado. Deben especificarse las responsabilidades del operador de la
planta de radiación (de emplear la dosis correcta, por ejemplo).
18.62 La dosis de radiación debe ser medida durante el
procedimiento de radiación. Con este fin, se deben emplear dosímetros que sean
independientes de la tasa de radiación, que indiquen una medida cuantitativa de
la dosis recibida por el producto mismo. Los dosímetros deben insertarse en la
carga en número adecuado y suficientemente cercanos unos a otros para asegurar
que haya un dosímetro en la cámara en todo momento. Cuando se trata de
dosímetros plásticos, deben emplearse dentro del tiempo límite después de su
calibración. Deben verificarse las absorbancias del dosímetro poco después de
su exposición a la radiación. Los indicadores biológicos pueden emplearse
solamente como un control adicional. Los discos de colores sensibles a la
radiación pueden usarse para distinguir entre los envases que han sido
sometidos a la radiación y aquellos que no; dichos discos no son indicadores de
una esterilización adecuada. La información obtenida debe formar parte del
registro del lote.
18.63 En los procedimientos de validación se debe asegurar
que se tengan en cuenta los efectos de las variaciones en la densidad de los
envases.
18.64 Los materiales deben manipularse de tal forma que se
evite la confusión entre los materiales que han sido irradiados y los que no.
Cada recipiente debe contar con un sensor de radiación que indique que ha sido
sometido al tratamiento con radiación.
18.65 La dosis total de radiación debe administrarse
dentro de un lapso preestablecido.
Esterilización por gases y productos fumigantes
18.66 Este método debe utilizarse únicamente cuando ningún
otro método es viable.
18.67 Diversos gases y productos fumigantes pueden
emplearse para la esterilización (ej. óxido de etileno, vapor de peróxido de
hidrógeno, etc). Durante el procedimiento de validación debe demostrarse que el
gas no produce ningún efecto nocivo para el producto y que las condiciones y el
tiempo asignado a la desgasificación son suficientes para reducir el gas
residual y los productos de reacción hasta límites aceptables definidos para el
tipo de producto o material. Dichos límites deben ser incorporados a las
especificaciones.
18.68 Es esencial el contacto entre el gas y los
microorganismos; deben tomarse precauciones para evitar la presencia de
organismos que puedan estar ocluidos en materiales tales como cristales o
proteína seca. La naturaleza y cantidad de los materiales de envasado pueden
influir significativamente en el proceso.
18.69 Antes de su exposición al gas, debe establecerse un
equilibrio entre los materiales y la humedad y temperatura requeridas por el
proceso. El tiempo empleado en esta operación debe considerarse en relación con
la necesidad de minimizar el tiempo transcurrido antes de la esterilización.
18.70 Cada ciclo de esterilización debe ser controlado
mediante indicadores biológicos, utilizando un número adecuado de unidades
distribuidas en toda la carga. La información obtenida por este medio debe
integrar el registro del lote.
18.71 Los indicadores biológicos deben ser almacenados y
usados de conformidad con las instrucciones del fabricante y su desempeño debe
ser verificado mediante controles positivos.
18.72 Para cada ciclo de esterilización deben mantenerse
registros del tiempo empleado para completar el ciclo, de la presión, de la
temperatura y de la humedad dentro de la cámara durante el proceso, como
también de la concentración del gas. La presión y la temperatura deben
registrarse en una tabla durante todo el ciclo. Estos datos deben formar parte
del registro del lote.
18.73 Después de la esterilización, la carga debe ser
almacenada en forma controlada y con la debida ventilación, para permitir que
el gas residual y los productos de reacción disminuyan hasta el nivel definido.
Este proceso debe validarse.
ESTERILIZACION POR FILTRACION
Filtración de productos farmacéuticos que no pueden ser
esterilizados en su envase final
18.74 Siempre que sea posible, los productos deben ser
esterilizados en el envase final, por esterilización térmica. Ciertas
soluciones y líquidos que no pueden ser esterilizados en el envase final,
pueden ser filtrados a través de un filtro estéril con poros de tamaño nominal
de 0,22 µm (o menos), o de uno que tenga características equivalentes de
retención de microorganismos, y cargados en envases previamente esterilizados.
Mediante tales filtros pueden eliminarse bacterias y hongos, pero no todos los
virus y micoplasmas. Se debe tener en cuenta la posibilidad de complementar el
proceso de filtración con algún grado de tratamiento térmico.
18.75 Debido a los potenciales riesgos adicionales que podría
significar el empleo del método de filtración, a diferencia de otros métodos de
esterilización, es aconsejable emplear un filtro de doble capa de filtración o
efectuar una segunda filtración con otro filtro que retenga microorganismos,
inmediatamente antes del llenado. La filtración final esterilizante debe
llevarse a cabo inmediatamente antes del llenado y lo más cerca posible al
punto de llenado.
18.76 No deben emplearse filtros que desprenden fibras. El
uso de filtros que contienen asbestos debe descartarse totalmente.
18.77 Debe controlarse la integridad del filtro empleando
un método apropiado, tal como la prueba de punto de burbuja, inmediatamente
después de cada uso (también es conveniente verificar el filtro de esta manera
antes del uso). El tiempo requerido para filtrar un volumen conocido de
solución a granel y la diferencia de presión a ser empleada a lo largo de la
filtración deben determinarse durante la validación y, si existen diferencias
significativas, éstas deben consignarse en el registro del lote.
18.78 No debe usarse el mismo filtro durante más de un día
de trabajo, a menos que se compruebe la inocuidad del uso adicional.
18.79 El filtro debe ser de naturaleza tal que no afecte
al producto adsorbiendo alguno de sus ingredientes o cediendo sustancias.
Personal
18.80 Sólo el número mínimo necesario de personal debe
estar presente en las áreas limpias; esto es especialmente importante durante
los procesos asépticos. De ser posible, las inspecciones y los controles deben
efectuarse desde afuera de las áreas respectivas.
18.81 Todos los empleados (incluyendo el personal de
limpieza y mantenimiento) que trabajan en dichas áreas deben someterse
regularmente a programas de capacitación en disciplinas relacionadas con la
correcta fabricación de productos estériles, incluyendo la higiene y
conocimientos básicos de microbiología. En caso de que sea necesario el ingreso
a las áreas de personas que no hayan recibido dicha capacitación (por ejemplo,
personal de mantenimiento contratado), deben ser capacitadas y supervisadas
cuidadosamente.
18.82 El personal que haya estado involucrado en el
procesado de materiales de tejidos animales o de cultivos de microorganismos
distintos de los usados en el proceso de fabricación en curso no debe ingresar
a las áreas de preparación de productos estériles, a menos que se hayan
aplicado procedimientos rigurosos y claramente definidos de decontaminación.
18.83 Deben mantenerse elevados niveles de higiene y
limpieza personal y los empleados involucrados en la fabricación de
preparaciones estériles deben ser instruidos para que informen cualquier
situación que pueda causar la liberación de cantidades o tipos anormales de
contaminantes; es conveniente que se efectúen exámenes periódicos para
determinar si existen dichas condiciones. Una persona competente designada
especialmente debe responsabilizarse de decidir acerca de las medidas que deban
adoptarse con respecto al personal que podría estar causando situaciones anormales
de peligro microbiológico.
18.84 A las áreas limpias no deben ingresar personas que vistan
ropas de calle y el personal que ingresa a los vestuarios deberá ya estar
vestido con la ropa protectora que se utiliza dentro de la fábrica. Con
respecto al cambio de ropas y al aseo personal, se deben seguir procedimientos
operativos normatizados.
18.85 El tipo de ropas y la calidad de las mismas dependen
del tipo de proceso de fabricación y del lugar de trabajo y las ropas deben
vestirse de tal forma que los productos estén protegidos de la contaminación.
18.86 Las personas que ingresan en las áreas limpias no
deben usar reloj de pulsera ni joyas, ni tampoco cosméticos de los cuales
puedan desprenderse partículas.
18.87 La vestimenta de las personas en el lugar de trabajo
debe ser acorde al grado del aire del área respectiva. A continuación se
describen las ropas exigidas para cada grado de aire:
- Grado D: El cabello y, cuando corresponda, la barba
deben cubrirse. Se deben usar ropas de protección y calzados o cubre calzados
apropiados. Deben adoptarse medidas apropiadas para evitar la contaminación
proveniente del exterior del área limpia.
- Grado C: El cabello y, cuando corresponda, la barba
deben cubrirse. Se deben usar trajes de una o dos piezas, cerrados en las
muñecas y con cuello alto y calzados o cubre calzados apropiados. De la
vestimenta empleada no debe prácticamente desprenderse fibra o partícula
alguna.
- Grado A/B: Una cofia debe cubrir totalmente el cabello
y, cuando corresponda, la barba; los bordes inferiores de la misma deben
meterse dentro del cuello del traje; debe usarse una máscara para evitar que la
cara desprenda gotas de sudor; deben usarse guantes de goma o material plástico
esterilizados que no estén recubiertos de talco, como también calzados
esterilizados o desinfectados; las botamangas de los pantalones deben meterse
dentro de los calzados y los extremos de las mangas deben meterse dentro de los
guantes. De la vestimenta empleada no debe prácticamente desprenderse fibra o
partícula alguna y debe retener toda partícula que se desprenda del cuerpo.
18.88 La vestimenta de calle no debe introducirse en los
vestuarios que llevan a las áreas de grado B y C. A cada empleado del área de
grado A/B se le debe suministrar vestimenta protectora limpia y esterilizada
para cada sesión de trabajo, o al menos una vez al día si los resultados del
control lo justifican. Los guantes deben desinfectarse regularmente durante las
operaciones y las máscaras y los guantes deben cambiarse para cada sesión de trabajo,
como mínimo. Es posible que sea necesario utilizar ropas descartables. Estas
medidas deben seguirse según procedimientos operativos normatizados.
18.89 La limpieza y el lavado de las ropas utilizadas en
las áreas limpias deben efectuarse de tal forma que no se les adhieran
partículas contaminantes que posteriormente puedan desprenderse de las mismas.
Es conveniente contar con instalaciones separadas para dichas ropas. Si las
ropas se deterioran debido a la limpieza o lavado inadecuados, puede aumentar
el riesgo de que de ellas se desprendan partículas. Las operaciones de lavado y
esterilización deben efectuarse de conformidad con procedimientos operativos
normatizados.
Locales
18.90 De ser posible, todos los locales deben diseñarse de
tal forma que se evite el ingreso innecesario de personal de supervisión o
control. El diseño de las áreas de grado B debe permitir que todas las
operaciones puedan ser observadas desde el exterior.
18.91 En las áreas limpias, todas las superficies
expuestas deben ser lisas, impermeables y sin grietas, para reducir al mínimo
el desprendimiento o la acumulación de partículas o microorganismos y permitir
la aplicación constante de sustancias limpiadoras y desinfectantes, donde sea
apropiado.
18.92 Para reducir la acumulación de polvo y para
facilitar la limpieza, no debe haber lugares que no puedan limpiarse y los
locales deben tener un mínimo número de repisas, estantes, anaqueles y equipos.
Las puertas deben estar construidas de tal forma que no tengan superficies que no
puedan limpiarse; por esta razón son inconvenientes las puertas corredizas.
18.93 En caso de existir cielo rasos falsos, éstos deben
cerrarse herméticamente para prevenir la contaminación proveniente del espacio
libre.
18.94 En la instalación de tuberías y conductos no deben
quedar huecos difíciles de limpiar.
18.95 Siempre que sea posible, se debe evitar la
instalación de sumideros y drenajes o bien excluirlos de las áreas donde se
efectúan operaciones asépticas. Donde haya necesidad de instalarlos, deben
diseñarse, ubicarse y mantenerse de tal manera que se reduzca al mínimo el
riesgo de contaminación microbiana; deben contar con sifones efectivos y
cierres de aire que sean eficientes y fáciles de limpiar, con el fin de
prevenir el reflujo de los líquidos.
18.96 Los vestuarios deben estar diseñados como esclusas
de aire, para separar las diferentes etapas del cambio de ropa, para minimizar
así la contaminación de las ropas de protección con microoganismos y
partículas. Dichas habitaciones deben limpiarse eficientemente con aire
filtrado. A veces es conveniente contar con vestuarios independientes para la
entrada y para la salida de las áreas limpias. Las instalaciones para el lavado
de las manos deben estar ubicadas solamente en los vestuarios, nunca en los
lugares donde se efectúan trabajos asépticos.
18.97 Las esclusas de aire no deben abrirse
simultáneamente. Se debe contar con un sistema de cierre interbloqueado y/o con
un sistema de alarma visual y/o auditivo para prevenir la apertura de más de
una puerta a la vez.
18.98 Una entrada de aire filtrado debe barrer eficazmente
el área y crear en todas las condiciones de trabajo, una presión positiva
respecto de todas las áreas adyacentes. Las áreas adyacentes de grados
diferentes deben tener un gradiente de presión de 10 – 15 pascales (valor
orientativo). Además, se debe prestar especial atención a la protección de la
zona de mayor riesgo, es decir, al ambiente inmediato al cual están expuestos y
con el cual toman contacto los productos y los componentes limpios. Es posible
que las recomendaciones concernientes al suministro de aire y a las diferencias
de presión tengan que ser modificadas, en caso de que sea necesario albergar
materiales tales como productos patogénicos, muy tóxicos, radioactivos, o virus
o bacterias vivos. Para algunas operaciones tal vez sea preciso contar con
instalaciones de descontaminación y de tratamiento del aire que sale de un área
limpia.
18.99 Debe demostrarse que los patrones de corriente de
aire no presenten riesgo de contaminación; así, por ejemplo, se debe tener
especial cuidado para asegurar que las corrientes de aire no distribuyan
partículas, provenientes de personas, máquinas u operaciones, hacia un área de
mayor riesgo para los productos.
18.100 Debe instalarse un sistema de advertencia que
indique cuando existe una falla en el suministro de aire. Entre una y otra
área, donde la diferencia de presión de aire es importante, debe instalarse un
indicador de presión y las diferencias deben registrarse regularmente.
18.101 Debe restringirse el acceso innecesario a las áreas
de llenado, como por ejemplo las zonas de llenado de grado A, donde podrían
colocarse barreras para tal efecto.
Equipos
18.102 No debe permitirse que una cinta transportadora
pase a través de una división colocada entre un área de grado B y un área de
menor grado de limpieza de aire, a menos que dicha cinta se someta a
esterilización continua (en un túnel de esterilización, por ejemplo).
18.103 Para el procesado de productos estériles deben
escogerse equipos, accesorios y servicios que puedan ser eficientemente
esterilizados por medio de vapor, calor seco u otros métodos.
18.104 Siempre que sea posible, el montaje y el
mantenimiento de los equipos debe realizarse de tal manera que las operaciones
de puedan llevarse a cabo fuera del área estéril. Si los equipos necesitan ser
esterilizados esto debe llevarse a cabo después del armado completo, si es
posible.
18.105 Cuando el mantenimiento de los equipos se efectúa
dentro de un área estéril, deben emplearse instrumentos y herramientas
estériles y el área debe ser esterilizada y desinfectada antes de volver a
iniciar el proceso.
18.106 Todos los equipos, incluyendo esterilizadores,
sistemas de filtración de aire y sistema de tratamiento de agua, incluso
destiladores, deben ser sometidos a un plan de mantenimiento y validación; debe
registrarse la autorización de uso otorgada después del mantenimiento de los
mismos.
18.107 Las plantas de tratamiento de agua deben ser
diseñadas, construidas y mantenidas de tal forma que se asegure la producción
confiable de agua de calidad apropiada. En su funcionamiento dichas plantas no
deben exceder la capacidad para la cual fueron diseñadas. En la producción,
almacenamiento y distribución se debe procurar impedir el crecimiento
microbiano, recurriendo, por ejemplo, a una circulación constante a una
temperatura superior a 70ºC o no más de 4ºC.
Sanitización
18.108 Es sumamente importante la sanitización de las
áreas limpias. Deben limpiarse frecuentemente, en forma completa, de
conformidad con un programa escrito aprobado. Los agentes sanitizantes
utilizados deben alternarse en forma periódico de acuerdo a un procedimiento
operativo normatizado. Deben efectuarse controles periódicos a fin de detectar
cepas de microorganismos resistentes. En vista de su limitada eficacia, la luz
ultravioleta no debe usarse en sustitución de la desinfección química.
18.109 Los desinfectantes y detergentes deben controlarse
para detectar su posible contaminación microbiana; sus diluciones deben
mantenerse en recipientes limpios y no deben ser guardadas por mucho tiempo a
no ser que hayan sido esterilizadas. Los desinfectantes y detergentes
utilizados en las áreas grado A y B deben ser esterilizados previamente a su
uso (Ver tabla 1). Si un recipiente está parcialmente vacío no debe rellenarse.
18.110 La fumigación de las áreas limpias puede ser útil
para reducir la contaminación microbiológica en los lugares inaccesibles.
Acabado de productos estériles
18.111 Los recipientes deben ser cerrados mediante métodos
debidamente validados. Los envases cerrados por fusión como, por ejemplo, las
ampollas de cristal o plástico deben someterse a una prueba de integridad del
100%. Los otros envases se deben muestrear según procedimientos operativos
normatizados para la prueba de integridad.
18.112 Los recipientes cerrados herméticamente al vacío
deben verificarse mediante el control de muestras de los mismos, para
establecer si el vacío se ha mantenido después de transcurrido un tiempo
predeterminado.
18.113 Los envases de productos parenterales llenos deben
inspeccionarse individualmente. Si la inspección es visual, debe efectuarse
bajo condiciones adecuadas y controladas de iluminación y fondo. Los operarios
que realizan la inspección deben someterse a controles periódicos de agudeza
visual, con lentes si los usan normalmente, y durante las inspecciones deben
tener descansos frecuentes. Si se utilizan otros métodos de inspección, éstos
deben validarse y los aparatos empleados deben ser controlados a intervalos
regulares. Los resultados deben quedar registrados.
Control de calidad
18.114 Las muestras tomadas para el ensayo de esterilidad
deben ser representativas de todo el lote, se deben incluir muestras tomadas de
las partes del lote que se considere de mayor riesgo de contaminación, por
ejemplo:
a. en el caso de productos que han sido llenados
asépticamente, entre las muestras se deben incluir las provenientes de envases
llenados al inicio y al final del lote y luego de alguna interrupción imprevista
del trabajo;
b. si se trata de productos que han sido esterilizados por
calor en sus envases finales, deben obtenerse muestras de la parte más fría de
la cámara.
18.115 El ensayo de esterilidad al que se somete el
producto terminado debe ser considerado sólo como la última de una serie de
medidas de control, mediante las cuales, se asegura la esterilidad. Sólo puede
interpretarse como parte de un conjunto que incluya los registros de las
condiciones ambientales y el procesado de los lotes. La esterilidad del
Producto Terminado se asegura por medio de la validación del ciclo de
esterilización en el caso de productos esterilizados en forma terminal, y por
medio del ensayo de "media-fills" realizado para productos que se
elaboran en forma aséptica. Se deben examinar, además, los registros del lote
procesado y los registros de calidad ambiental. El ensayo de esterilidad debe
ser validado para cada producto. Se deben usar los métodos codificados tanto
para la validación como para la realización del ensayo.
18.116 En aquellos casos en que la liberación paramétrica
ha sido autorizada por la Autoridad Sanitaria Nacional, se debe prestar especial atención a la validación y supervisión de todo el proceso de elaboración.
(Ver Anexo III)
18.117 Los lotes que no pasan la prueba de esterilidad no
pueden ser aprobados sobre la base de una segunda prueba, a menos que se lleve
a cabo una investigación del tipo de miroorganismo encontrado y de los
registros sobre las condiciones ambientales y el procesado de los lotes y como
resultado de la misma se demuestre que la prueba original no era válida. La
repetición del ensayo se debe realizar con el doble de muestras.
18.118 Para productos inyectables, tanto al agua para
inyectables como a los productos intermedios y terminados se les debe
investigar la presencia de endotoxinas, empleando el método establecido por la Farmacopea Argentina. Cuando una muestra no pasa la prueba, debe investigarse la causa y
adoptarse las medidas correctivas necesarias.
ANEXO I
Aplicación de la Metodología de Análisis de Peligros y Puntos Críticos de Control en la Producción de Medicamentos
1. INTRODUCCION
Esta metodología apunta a prevenir peligros conocidos y a
reducir los riesgos de su ocurrencia en puntos específicos. Este texto
proporciona lineamientos generales para el uso del sistema HACCP en el
aseguramiento de la calidad de medicamentos, aún cuando detalles de su
aplicación puedan variar según las circunstancias. Este anexo no provee
información detallada de los peligros principales.
Los procedimientos (entre los que se incluyen las BPF),
atienden las condiciones de operación y proveen las bases para el sistema
HACCP. El HACCP es un método sistemático para la identificación, valoración y
control de peligros que afecten la seguridad.
Además, el HACCP puede extender este concepto, incluyendo
un análisis de las variables críticas de la calidad al igual que una valoración
de los peligros que afectan la seguridad de los trabajadores y peligros de
contaminación del medio ambiente directamente relacionado a los procesos
concernientes (en particular en sistemas abiertos).
Las BPF para productos farmacéuticos requieren tanto la
validación de los procesos críticos como de los cambios en los procesos de
manufactura que pueden afectar la calidad del producto final. La experiencia
muestra que muchos procesos de manufactura contienen etapas que son críticas
desde el punto de vista de la variación en la calidad final.
El HACCP es una herramienta para valorar peligros y
establecer sistemas de control centrados en la prevención, en lugar de confiar
en acciones correctivas basadas en el control final del producto. Todo sistema
HACCP es capaz de adaptarse a cambios, tales como avances en diseño de equipos
y procesos productivos u otros desarrollos tecnológicos.
2. PRINCIPIOS
El sistema HACCP se basa en siete principios. Para la
aplicación de estos principios, se recomienda desarrollarlos en 14 etapas, las
cuales son propuestas en la sección 5.
Algunas etapas están relacionadas con principios
específicos, mientras que otras sirven como una introducción a los conceptos.
Los siete principios son:
1. Realizar un Análisis de Peligros
2. Determinar los Puntos Críticos de Control
3. Establecer los parámetros y límites críticos
4. Establecer un sistema de monitoreo de los PCC
5. Establecer las acciones correctivas a realizar cuando
el monitoreo indique que un PCC no está bajo control.
6. Establecer documentación concerniente a todos los
procesos y conservar los registros apropiados a esos principios y su aplicación.
7. Establecer procedimientos para verificar que el sistema
HACCP esta trabajando efectivamente.
3. REQUISITOS PREVIOS PARA LA APLICACION DEL SISTEMA HACCP
3.1 Los siguientes lineamientos deberían ser utilizados en
la aplicación del sistema HACCP:
• Antes que el sistema HACCP sea aplicado a un determinado
sector, este sector debe estar operando de acuerdo con los principios de las
BPF.
• Es necesario un comité de gestión de la calidad para
implementar un sistema HACCP.
• El HACCP debe ser aplicado a cada etapa específica
separadamente.
• Los PCC que son dados como un ejemplo determinado en
algún documento (incluido las BPF) puede no ser el único identificado para una
aplicación específica o puede ser de una naturaleza diferente.
• La implementación del HACCP debe ser revisada y
necesariamente cambiada cuando se realice alguna modificación en el producto,
proceso o etapa.
• En la implementación del HACCP es necesario tomar en
cuenta la naturaleza y el tamaño de la operación.
• El formato de cada plan HACCP puede variar, pero deben
ser preferentemente específicos para cada producto, proceso u operación en
particular. Los planes HACCP generales pueden servir como guías útiles en el
desarrollo de planes específicos para productos o procesos; sin embargo, es
esencial que las condiciones únicas dentro de cada uno de ellos sean
consideradas durante el desarrollo de todos los componentes del plan HACCP.
4. ENTRENAMIENTO Y CAPACITACION
4.1 Para la efectiva implementación de un plan HACCP, se
debe capacitar al personal en lo que respecta los principios y aplicación del
HACCP.
4.2 En el desarrollo del entrenamiento específico para
sustentar un plan HACCP, las instrucciones de trabajo y los procedimientos
deben formularse por escrito para definir las tareas del personal operativo
destinado a cada PCC. Los encargados de monitorear cada PCC deben recibir
entrenamiento específico.
4.3 La capacitación, información y entrenamiento debe ser
provista sobre el control de peligros en todas las etapas de producción y
abastecimiento.
4.4 El personal debe comprender que el HACCP está
implementado, y que el informarse es necesario para que este funcione de manera
adecuada, y también que los materiales y equipamientos necesarios deben ser
provistos para el control de los PCC.
4.5 Todo el entrenamiento y capacitación del personal que
interviene en el plan HACCP debe ser registrado.
5. APLICACION
5.1 Principio: La aplicación de los principios del HACCP
debe consistir de las siguientes etapas, como una secuencia lógica para la
aplicación del sistema HACCP.
5.2 Definir el alcance del Plan HACCP. Se debe definir el
alcance del plan HACCP y debe describir los segmentos de la producción
involucrados e identificar las clases de peligros a ser tratados.
5.3 Ensamblar un equipo HACCP. La elaboración de productos
farmacéuticos debe asegurar que el conocimiento y la experiencia de productos
específicos esté disponible para el desarrollo de un efectivo plan HACCP. Esto
puede ser alcanzado de mejor manera mediante la elección de un equipo
multidisciplinario. Los miembros del equipo deberían provenir de todas las
áreas relevantes, como ser investigación y desarrollo, producción, control de
calidad, aseguramiento de calidad, microbiología, ingeniería y distribución u
otras. Los miembros del equipo deben tener conocimientos específicos y
experiencia relacionada a los productos y los procesos. En las áreas donde no
se cuenta con experiencia, pueden ser incorporados asesores externos. Los
miembros del equipo deben ser capaces de:
(a) Realizar un análisis de peligros
(b) Identificar los peligros potenciales
(c) Identificar los peligros que deben ser controlados
(d) Recomendar controles y límites críticos
(e) Diseñar procedimientos de monitoreo y verificación
(f) Establecer las acciones correctivas apropiadas cuando
ocurra una desviación
(g) Verificar el plan HACCP
5.4 Describir los productos y procesos. Se debe realizar
una descripción total del producto y de los procesos involucrados, incluyendo
información relevante relacionada con la calidad como ser, la composición,
propiedades físico - químicas, estructura, pH, temperaturas, métodos de
limpieza, tratamientos bacteriológicos o bacteriostáticos (por ejemplo
esterilización por calor), secado, mezclado, combinación de fases,
acondicionamiento y condiciones de almacenamiento. El método de distribución y
transporte también debe ser descripto, especialmente cuando los productos son
termolábiles.
5.5 Identificar de la intención de uso. La intención de
uso debe estar basada en la expectativa de uso por parte del consumidor final.
Deben ser considerados casos específicos como ser grupos vulnerables, por
ejemplo, pacientes geriátricos, pacientes pediátricos e inmunosuprimidos.
5.6 Construir un diagrama de flujo. El diagrama de flujo
debe ser construido por el equipo HACCP, y debe cubrir todas las operaciones y
decisiones del proceso. Cuando el sistema HACCP es aplicado a una etapa
específica, se deben considerar la etapa anterior y la posterior a esta
operación. Un diagrama de flechas y cuadros puede ser suficientemente
descriptivo.
5.7 Confirmar el diagrama de flujo. El equipo HACCP debe
confirmar el diagrama de flujo en comparación con las operaciones de producción
durante todos los estadios y tiempos de operación. Las modificaciones al
diagrama de flujo deben ser realizadas cuando sea apropiado, lo cual debe
quedar registrado.
5.8 Realizar un listado de todos los peligros potenciales
asociados a cada etapa, realizar un análisis de riesgos y considerar alguna
medida de control para los peligros identificados. (Principio 1). Cuando se
realiza el análisis de riesgo, los peligros relacionados con la seguridad deben
ser distinguidos de los peligros concernientes a la calidad.
5.8.1 El equipo HACCP debe listar todos los peligros que
pueden ser razonablemente esperados a ocurrir en cada etapa consideradas de la
elaboración, control, depósito y distribución.
5.8.2 Se debe realizar un análisis de peligro para
identificar que, para los peligros de cada naturaleza, se elimine o reduzca su
riesgo a un nivel aceptable. Se requiere un minucioso análisis de peligros para
asegurar un efectivo punto de control. Se recomienda dos etapas para el
análisis de peligros.
(a) Durante la primera etapa el equipo debería revisar los
materiales, actividades, equipamiento, acondicionamiento, distribución e
intención de uso del producto. Debe ser diseñada una lista de los potenciales
peligros (biológicos, químicos y físicos) que pueden ser introducidos,
incrementados o controlados en cada etapa.
Cuando sea posible, lo siguiente debe ser incluido en el
análisis de peligros:
- La probable ocurrencia de peligros y la severidad de sus
efectos adversos a la salud
- La evaluación cualitativa/cuantitativa de la presencia de
los peligros
- La supervivencia o multiplicación de microorganismos de
preocupación
- La producción o persistencia en drogas de toxinas,
químicos o agentes físicos
- Las condiciones principales para lo anteriormente dicho
(b) Durante la segunda etapa, debe realizarse una
evaluación de los riesgos, donde deben ser estimada la severidad de los
peligros potenciales y la probabilidad de su ocurrencia. El equipo debe luego
decidir que peligros potenciales deben ser tratados en el plan HACCP, y que
medidas de control pueden ser aplicadas, si existe, para cada peligro. Más de
una medida de control puede ser requerida para controlar un peligro específico
y más de un peligro puede ser controlado mediante una medida de control
específica. Deben ser considerados, como mínimo, peligros potenciales en
relación a:
- Materiales e ingredientes
- Características físicas y composición del producto
- Procedimientos productivos
- Instalaciones
- Equipamiento
- Acondicionamiento
- Sanitización e higiene
- Personal
- Riesgos de explosión
- Confusión
5.9 Determinar los Puntos Críticos de Control. (Principio
2). Se deben determinar los Puntos de Control que sean Críticos para cada
etapa, proceso o segmento de la línea productiva considerada. Un PCC en el
sistema HACCP puede ser más fácilmente determinado mediante el uso de un árbol
de decisiones, que facilita un abordaje lógico. El modo que un árbol de
decisiones es usado podría depender de la operación involucrada, por ejemplo,
producción, acondicionamiento, almacenamiento o distribución. Se debe dar
entrenamiento en el uso del árbol de decisiones. Si un peligro fue identificado
en una etapa donde el control es necesario para la seguridad, y la medida de
control no existe en esta etapa, o en alguna otra, el producto o el proceso
debe ser modificado en esta etapa, o en una etapa anterior o posterior,
incluyendo en tal una medida de control.
5.10 Establecer los parámetros y límites críticos para
cada PCC. (Principio 3). Los límites críticos deben ser especificados y verificados
para cada punto critico de control. Más de un límite crítico puede, algunas
veces, ser establecido para una etapa en particular. Con frecuencia el criterio
utilizado incluye mediciones de temperatura, tiempo, nivel de humedad, pH, y
parámetros sensoriales tales como apariencia y textura. Los límites críticos
deben estar basados científicamente.
5.11 Establecer un sistema de monitoreo para cada PCC.
(Principio 4). El monitoreo es el proceso de mediciones u observaciones de un
PCC relativo a sus límites críticos. El monitoreo debe estar establecido y ser
registrado.
5.11.1 El procedimiento de monitoreo utilizado debe ser
capaz de detectar una pérdida de control de un PCC, y esta información debe
estar disponible en tiempo para realizar ajustes, para asegurar el control del
proceso y prevenir violaciones a los límites críticos. Cuando sea posible, se
debe realizar modificaciones del proceso, cuando los resultados del monitoreo
indican una tendencia hacia la pérdida de control de un PCC. Estos ajustes deben
ser realizados antes que ocurra la desviación.
5.11.2 Los datos derivados del monitoreo deben ser
evaluados por una persona asignada, y con el conocimiento y autoridad
suficiente para llevar a cabo acciones correctivas cuando lo indique.
5.11.3 Si el monitoreo es discontinuo, la cantidad o la
frecuencia de las mediciones deben ser las suficientes para garantizar que el
PCC está bajo control. Muchos procesos de monitoreo para PCC podrían necesitar
ser realizados rápidamente porque ellos se relacionan a procesos en línea y
éstos no podrían ser largos ensayos analíticos. Por esta razón, mediciones
físicas y químicas son frecuentemente preferidas a ensayos microbiológicos
porque ellas pueden ser realizadas rápidamente y pueden frecuentemente dar idea
del estado microbiológico del producto.
5.11.4 El personal que realiza el monitoreo de PCC y las
medidas de control, deben ser parte del área o departamento producción (por ej.
supervisores de línea, personal de mantenimiento) y, cuando sea apropiado,
personal de control de calidad. Ellos deben ser entrenados en procesos de
monitoreo.
5.11.5 Cuando se utilice un monitoreo continuo, se debe
establecer su frecuencia de registro y la recolección de datos debe realizarse
de manera estadística o por un sistema de muestreo.
5.11.6 Todos los registros y documentos asociados con el
monitoreo de PCC deben ser firmados y fechados mediante la(s) persona(s) que
lleva a cabo el monitoreo y revisados por el responsable correspondiente.
5.12 Establecer acciones correctivas (Principio 5). Se
deben desarrollar acciones correctivas específicas para cada PCC para ser
implementadas cuando ocurre una desviación de los límites críticos.
5.12.1 Las acciones correctivas deben asegurar que el PCC
vuelva a estar bajo control. Las acciones correctivas deben incluir, al menos,
lo siguiente:
(a) Determinación y corrección de la causa del
incumplimiento;
(b) Determinación del destino del producto fuera de
límites;
(c) Registro de la(s) acción(es) correctiva(s) que ha(n)
sido tomada(s);
5.12.2 Las acciones correctivas específicas deben estar
definidas por adelantado para cada PCC y estar incluidas en el plan de HACCP.
Como mínimo, el plan HACCP debe especificar: que se debe hacer cuando ocurre
una desviación, quien es responsable de ejecutar las acciones correctivas, y
que el registro de las acciones tomadas sea guardado y mantenido. Se les debe
asignar la responsabilidad de la aplicación de acciones correctivas a los
individuos que tienen una comprensión cuidadosa del proceso, del producto y del
plan de HACCP.
5.12.3 Los procedimientos de la disposición del producto y
las desviaciones deben ser documentadas en los registros del plan HACCP.
5.13 Establecer un sistema de registro y documentación
(Principio 6). Se debe establecer un sistema de registro y documentación
eficiente y adecuado, lo cual es esencial para la aplicación de un sistema
HACCP, y debe ser apropiado para la naturaleza y magnitud de la operación.
Se debe documentar, como mínimo, las siguientes
actividades:
• Análisis de Riesgos;
• Determinación de los PCC;
• Plan HACCP;
• Determinación de los límites críticos;
Se debe registrar, como mínimo, las siguientes
actividades:
• Monitoreo de los PCC;
• Etapas procesadas;
• Peligros asociados;
• Límites críticos;
• Procedimientos y listados de verificación;
• Desviaciones;
• Acciones correctivas asociadas;
• Modificaciones al sistema HACCP;
5.14 Revisar el plan HACCP. Se debe realizar una revisión
inicial del plan de HACCP para determinar si se han sido identificados todos
los peligros y, si el plan HACCP se ejecuta correctamente, estos peligros sean
controlados con eficacia.
5.15 Establecer los procedimientos de verificación
(Principio 7). Se debe establecer procedimientos para verificar que el sistema
HACCP implementado es efectivo.
5.15.1 Se pueden utilizar, para determinar si el sistema
de HACCP está trabajando correctamente, los métodos de verificación y
auditoría, procedimientos y ensayos, incluyendo el muestreo aleatorio y su
análisis. La frecuencia de la verificación debe ser suficiente para confirmar
el funcionamiento apropiado del sistema HACCP. Ejemplos de las actividades de
la verificación incluyen:
• Revisión del sistema de HACCP y de sus registros;
• Revisión de desviaciones y la disposiciones del producto;
• Confirmación que los PCCs son mantenidos bajo control.
5.15.2 La revisión de la información para verificar el
plan de HACCP debe incluir:
(a) Estudios científicos;
(b) Observaciones, mediciones y evaluaciones en planta.
Por ejemplo, para la verificación del proceso de esterilización térmica por
calor húmedo de inyectables estériles, debe incluir la justificación científica
de una destrucción apropiada de los microorganismos patógenos y que los
estudios de los tiempos de calentamiento, presión y temperaturas, son
necesarias para confirmar que las condiciones de esterilización aseguren que la
carga entera esté mantenida a la temperatura requerida por el tiempo requerido.
5.15.3 Las verificaciones posteriores se deben realizar y
documentar por el equipo HACCP. Las verificaciones deben hacerse cuando hay un
fallo inexplicable del sistema, cuando ocurre un cambio significativo en el
producto, proceso o acondicionamiento, o cuando se reconocen nuevos peligros.
5.15.4 Se debe realizar una evaluación comprensiva
periódica del sistema por una parte imparcial o terceros independientes del
plan HACCP. Esto debe incluir una evaluación técnica del análisis de peligro y
de cada elemento del plan HACCP, así como una revisión en el sitio de todos los
diagramas flujo y los registros apropiados de las operaciones del plan. Esta
verificación comprensiva es independiente de los otros procedimientos de
verificación y se debe realizar para asegurar que el plan HACCP da como
resultado el control de los todos los peligros.
5.15.5 Si los resultados de la verificación comprensiva
identifican deficiencias, el equipo HACCP debe modificar el plan HACCP como sea
necesario.
5.15.6 Los individuos que realicen la verificación deben
tener un criterio técnico apropiado para realizar esta función. En lo posible,
la verificación debe incluir acciones para confirmar la eficacia de todos los
elementos del plan HACCP.
GLOSARIO
Acción correctiva
Cualquier acción a ser tomada cuando los resultados del
monitoreo de un PCC indican una pérdida de control.
Análisis de Peligro
El proceso de recolección y evaluación de información que
debe ser realizado en el plan HACCP.
Control
El estado en el cual se han seguido los procedimientos
correctos y en el cual se están cumpliendo los criterios establecidos.
Controlar
Realizar todas las acciones necesarias para asegurar el
cumplimiento de los criterios establecidos en un plan HACCP.
Desviación
El no cumplimiento de un límite crítico.
Diagrama de flujo
Representación sistemática de la secuencia de pasos u
operaciones involucradas en la fabricación de un medicamento.
Límite crítico
Criterio que separa la aceptabilidad de la
inaceptabilidad.
Medida de control
Cualquier acción y actividad que puede ser usada para
prevenir o eliminar un peligro que pueda afectar la calidad del medicamento o
reducir el riesgo a un nivel aceptable.
Monitoreo
El acto de conducir una secuencia planeada de
observaciones o mediciones de parámetros de control para reconocer si un PCC
está bajo control.
Plan HACCP
Documento preparado según los principios del HACCP para
asegurar, en un segmento de la línea productiva, el control de los riesgos que
son significativos para la calidad del medicamento.
Punto Crítico de Control (PCC)
Paso en el cual se puede aplicar el control, y que es
esencial para prevenir o eliminar un peligro que puede afectar la calidad del
medicamento o reducir el riesgo a un nivel aceptable.
Peligro
Cualquier circunstancia en la producción, control y
distribución de medicamentos que puede causar un efecto adverso para la salud o
un desvío de la calidad.
Validación
Colección y evaluación de datos, provenientes del proceso
de etapas de desarrollo y continuando hacia las fases de producción, que
asegura que los procesos de elaboración (incluyendo equipamiento,
instalaciones, personal y materiales) son capaces de proporcionar los
resultados esperados en forma consistente y continua. Validación es el
establecimiento de evidencia documentada que un sistema hace lo que tiene que
hacer.
Verificación
La aplicación de métodos, ensayos, controles y otras
evaluaciones, además del monitoreo, para determinar el cumplimiento del plan
HACCP.
ANEXO II
Calificación y validación
1. PRINCIPIO
El presente anexo describe los principios de calificación
y validación aplicables a la fabricación de medicamentos. Las Buenas Prácticas
de Fabricación exigen que los fabricantes identifiquen las tareas de validación
que son necesarias para demostrar el control de los aspectos críticos de sus operaciones
en particular. Se debe validar todo cambio significativo en las instalaciones,
equipos y procesos que pueda influir en la calidad del producto. Debe emplearse
un enfoque de análisis de riesgo para determinar el ámbito y la amplitud de la
validación.
2. PLANIFICACION DE LA VALIDACION
2.1 Todas las actividades de validación deben estar
planificadas. Los elementos clave de un programa de validación deben estar
definidos y documentados con claridad en un plan maestro de validación (PMV).
2.2 El PMV de validación debe ser un documento resumido,
es decir, breve, conciso y claro.
El PMV debe contener, como mínimo, datos sobre los
siguientes aspectos:
(a) Manual de calidad
(b) Estructura organizativa de las actividades de
validación.
(c) Resumen de instalaciones, sistemas, equipos y procesos
que se deben validar.
(d) Formato de la documentación: el formato que se emplea
para los protocolos e informes.
(e) Planificación y calendario.
(f) Control de cambios.
(g) Referencia a documentos anteriores.
2.4 Cuando se trate de proyectos grandes, puede ser
necesario crear planes maestros de validación independientes.
3. DOCUMENTACION
3.1 Se debe elaborar un protocolo por escrito en el que se
especifique el método de calificación y validación. Este protocolo debe ser
revisado y aprobado. Además, deben estar especificadas las etapas fundamentales
y los criterios de aceptación.
3.2 Se debe preparar un informe que establezca la relación
entre el protocolo de calificación y/o el de validación, que resumirá los
resultados obtenidos, comentará las desviaciones observadas y extraerá las
conclusiones pertinentes, incluidas las recomendaciones sobre cambios
necesarios para corregir las deficiencias. Todo cambio, en el plan tal como se
ha definido en el protocolo, debe documentarse con la justificación
correspondiente.
3.3 Una vez llevada a cabo una calificación satisfactoria,
se debe efectuar una aprobación formal para la siguiente fase de la
calificación y validación en forma de autorización por escrito.
4. CALIFICACION
Calificación del diseño
4.1 El primer elemento de la validación de nuevas
instalaciones, sistemas o equipos podría ser la calificación del diseño (CD).
4.2 Se debe demostrar y documentar la adecuación del
diseño a las Buenas Prácticas de Fabricación.
Calificación de la instalación
4.3 La calificación de la instalación (CI) se debe
realizar en caso de instalaciones, sistemas y equipos nuevos o modificados.
La calificación de la instalación debe incluir, entre
otras cosas, lo siguiente:
a) comprobación de la instalación de equipos, conductos,
servicios e instrumentos respecto a diagramas y especificaciones actualizadas
de ingeniería;
b) recopilación y verificación de las instrucciones de
operación y funcionamiento del proveedor y de las exigencias de mantenimiento;
(c) requisitos de calibración;
(d) verificación de los materiales de construcción.
Calificación de operación
4.5 La calificación del funcionamiento (CO) debe
realizarse tras la calificación de la instalación.
4.6 La calificación del operación debe incluir, entre
otras cosas, lo siguiente:
a) ensayos que han sido desarrollados a partir del
conocimiento sobre procesos, sistemas y equipos;
b) ensayos que incluyan una situación o un conjunto de situaciones
que abarquen los límites máximos y mínimos del trabajo, estas condiciones
denominadas con frecuencia como "caso más desfavorable".
4.7 La finalización de forma satisfactoria de la
calificación de operación debe permitir concluir con los procedimientos de
calibración, fabricación y limpieza, la capacitación de los operarios y las
exigencias de mantenimiento preventivo. Así, permitirá la aprobación formal de
las instalaciones, sistemas y equipos.
Calificación del proceso (CP)
4.8 La calificación del proceso (CP) debe efectuarse una
vez realizadas satisfactoriamente la calificación de la instalación y la
calificación del operación.
La calificación del proceso debe incluir, entre otras
cosas, lo siguiente:
a) ensayos, empleando materiales de producción,
componentes sustitutivos calificados o simulaciones de productos, que se hayan
desarrollado a partir del conocimiento sobre los procesos y las instalaciones,
sistemas o equipos;
b) ensayos que incluyan una situación o un conjunto de
situaciones que abarquen los límites máximos y mínimos de operación.
4.9 Aunque la calificación de resultados se describe como
una actividad independiente, en ciertos casos será apropiado realizarla junto
con la calificación de operación.
Calificación de las instalaciones, sistemas y equipos
establecidos (en uso)
4.10 Se deben ofrecer pruebas que respalden y verifiquen
los parámetros y límites de operación para las variables críticas del equipo.
Además, se debe documentar la calibración, la limpieza, el mantenimiento preventivo,
los procedimientos de fabricación y los procedimientos y protocolos de
capacitación de los operarios.
5. VALIDACION DE PROCESOS
Generalidades
5.1 Las exigencias y principios mencionados en este
capítulo son aplicables a la fabricación de formas farmacéuticas. Cubren la
validación inicial de procesos nuevos, la validación posterior de procesos
modificados y la revalidación.
5.2 La validación de procesos normalmente se debe
completar antes de la distribución y venta del medicamento (validación prospectiva).
En circunstancias excepcionales que impidan dicha validación, será necesario
validar los procesos durante la producción sistemática (validación
concurrente). Asimismo deberán validarse los procesos de aquellos producto que
ya se han comercializado, basada en datos acumulados de fabricación, ensayos y
lotes de control (validación retrospectiva).
5.4 Las instalaciones, sistemas y equipos que se vayan a
utilizar deben estar calificados previamente y las técnicas analíticas estar
validadas. Además, el personal que participe en las tareas de validación debe
haber recibido la capacitación apropiada, la cual debe estar debidamente
documentada.
5.5 Las instalaciones, sistemas, equipos y procesos deben
ser evaluados periódicamente para verificar que su funcionamiento sigue siendo
válido.
Validación prospectiva
5.6 La validación prospectiva debe incluir, entre otras
cosas, lo siguiente:
(a) breve descripción del proceso;
(b) resumen de las fases críticas del proceso de
fabricación que se van a investigar;
(c) listado de los equipos o instalaciones que se van a
utilizar (incluido el equipo de medición, control o registro) junto con su
estado de calibración;
(d) especificaciones del producto terminado para su
aprobación;
(e) listado de métodos analíticos, según corresponda;
(f) propuesta de controles durante el proceso, junto con
sus criterios de aceptación;
(g) ensayos adicionales, junto con sus criterios de
aceptación y la validación analítica;
(h) plan de muestreo;
(i) métodos de registro y evaluación de los resultados;
(j) funciones y responsabilidades;
(k) calendario propuesto.
5.7 Mediante el proceso así definido, incluidos los
componentes especificados, se debe elaborar una serie de lotes del producto
final en condiciones de rutina. En teoría, el número de repeticiones del
proceso y de las observaciones realizadas deberá bastar para que se pueda
establecer el margen normal de variación y las tendencias para facilitar datos
suficientes para su evaluación. Se considera como validación aceptable para el
proceso no menos de tres lotes o repeticiones consecutivas que cumplan con los
parámetros especificados.
5.8 Los lotes realizados para la validación del proceso
deben ser del mismo tamaño que los lotes previstos a escala industrial.
5.9 Si los lotes empleados para la validación están
destinados a su venta o suministro, las condiciones de producción deben cumplir
plenamente las exigencias de las Buenas Prácticas de Fabricación, incluido el
resultado satisfactorio del ejercicio de validación, y las de autorización de
comercialización.
Validación concurrente
5.10 La decisión de recurrir a la validación concurrente
debe estar justificada, documentada y debe contar con la aprobación del
personal autorizado.
5.11 Las exigencias de documentación para la validación
concurrente son las mismas especificadas para la validación prospectiva.
Validación retrospectiva
5.12 La validación retrospectiva solamente es aceptable
para procesos ya establecidos y será inadecuada cuando se han producido cambios
recientes en la composición del producto, en los procedimientos de elaboración
o en el equipamiento.
5.13 La validación de estos procesos debe basarse en datos
históricos. Sus fases incluyen la preparación de un protocolo específico y el
registro de los resultados de la revisión de los datos, de los que se extraerá
una conclusión y una recomendación.
5.14 Los datos empleados para esta validación se deben
extraer, entre otras fuentes, de los archivos de elaboración y
acondicionamiento de lotes, diagramas de control de proceso, cuadernos de
mantenimiento, registros de cambios de personal, estudios de adecuación de
proceso, datos sobre productos terminados, incluidas las fichas de tendencias,
y resultados de los estudios de estabilidad.
5.15 Los lotes seleccionados para la validación
retrospectiva deben ser representativos de todos los lotes fabricados durante
el período de revisión, incluidos los que no cumplan las especificaciones, y su
número será suficiente para demostrar la consistencia del proceso. Quizá sea
preciso efectuar ensayos adicionales en muestras conservadas para obtener la
cantidad o el tipo de datos necesarios para validar el proceso
retrospectivamente.
5.16 A efectos de validación retrospectiva, generalmente se
deben examinar datos de entre 10 y 30 lotes consecutivos para evaluar la
consistencia del proceso.
6. VALIDACION DE LA LIMPIEZA
6.1 Se debe efectuar la validación de la limpieza a fin de
confirmar la eficacia de los procedimientos de limpieza. La selección de
límites para residuos de productos, agentes de limpieza y contaminación
microbiana, debe estar justificada razonablemente, según los materiales
empleados. Los límites deben ser posibles de alcanzar y de verificar.
6.2 Se deben utilizar métodos analíticos validados lo
bastante sensibles para detectar residuos o contaminantes. El límite de
detección para cada método analítico deberá ser de la sensibilidad suficiente
para detectar el nivel aceptable establecido del residuo o contaminante.
6.3 Normalmente sólo es preciso validar procedimientos de
limpieza para las superficies del equipo que entren en contacto con los
productos, pero deben tenerse en cuenta las partes que no entren en contacto
directo. Se deben validar los intervalos entre el uso del equipo y su limpieza,
así como entre ésta y la reutilización del mismo. Se deben determinar los
intervalos de limpieza así como los métodos empleados.
6.4 En caso de procedimientos de limpieza para productos y
procesos similares, se considera aceptable seleccionar una gama representativa
de productos relacionados. Se podrá realizar un único estudio de validación que
siga el método del "caso más desfavorable" y tenga en cuenta los
aspectos críticos.
6.5 Se deben efectuar no menos de tres aplicaciones
consecutivas del procedimiento de limpieza con resultados satisfactorios para
demostrar que el método está validado.
6.6 El método de "ensayar hasta que esté limpio"
no se considera una alternativa apropiada para la validación de la limpieza.
7. CONTROL DE CAMBIOS
7.1 Debe existir procedimientos operativos normatizados
para describir las acciones que se deben realizar si se propone un cambio en
una materia prima, el componente de un producto, un equipo del proceso, el
entorno (o la instalación) de fabricación, el método de elaboración o ensayo, o
cualquier otro cambio que pueda afectar a la calidad del producto o la
reproducibilidad del proceso. Los procedimientos de control de cambios
garantizarán que se generan datos justificativos suficientes para demostrar que
el proceso revisado dará como resultado un producto de la calidad deseada, de
acuerdo con las especificaciones aprobadas.
Los cambios que afecten las condiciones bajo las cuales se
concedió la autorización de comercialización deberán ser aprobados por la Autoridad Sanitaria Nacional.
7.2 Todos los cambios que puedan influir en la calidad o
reproducibilidad del proceso, o modifiquen las condiciones bajo las cuales se
concedió la autorización de comercialización deben solicitarse, documentarse y
aceptarse formalmente. Se debe evaluar y se realizar un análisis de riesgo de
los posibles efectos que originaría un cambio de instalaciones, sistemas o
equipos sobre el producto. Se debe determinar la necesidad de una nueva
calificación y revalidación, y la amplitud de las mismas.
8. REVALIDACION
8.1 Las instalaciones, sistemas, equipos y procesos,
incluida la limpieza, deben ser evaluados periódicamente para confirmar que
siguen siendo válidos. Cuando no se hayan producido cambios significativos
respecto al estado validado, esta necesidad de revalidación se cubrirá con una
revisión que demuestre que las instalaciones, sistemas, equipos y procesos
cumplen las exigencias obligatorias.
GLOSARIO
A continuación se incluyen las definiciones de términos
relacionados con la calificación y validación que no se ofrecen en el glosario
de las Buenas Prácticas de fabricación, pero que se utilizan en el presente
anexo.
Análisis de riesgos
Método destinado a evaluar y clasificar los parámetros
fundamentales de la funcionalidad de un equipo o proceso.
Calificación de la instalación
Verificación documentada de que los locales, sistemas y
equipos, tal como se han instalado o modificado, se ajustan al diseño aprobado
y a las recomendaciones del fabricante.
Calificación del diseño
Verificación documentada de que el diseño propuesto para
los locales, sistemas y equipos es adecuado al propósito para el que están
destinados.
Calificación de resultados
Verificación documentada de que los locales, sistemas y
equipos, de la manera en que están conectados entre ellos, pueden ofrecer
resultados eficaces y reproducibles basados en el método de elaboración
aprobado y en las especificaciones del producto.
Calificación de operación
Verificación documentada de que los locales, sistemas y
equipos, tal como se han instalado o modificado, funcionan de la manera
esperada en todas las circunstancias de funcionamiento previstas.
Caso más desfavorable
Condición o conjunto de condiciones que abarquen los
límites máximos y mínimos de elaboración, así como las circunstancias, dentro
de los procedimientos de elaboración normatizados, que plantean mayores
posibilidades de fallos para el producto o proceso en comparación con las
condiciones ideales. Estas condiciones no provocan necesariamente fallos en el
producto o proceso.
Control de cambios
Un sistema formal por el cual representantes calificados
de las disciplinas apropiadas revisan los cambios propuestos o efectuados que
puedan influir en el estado validado de los locales, sistemas, equipos o
procesos. Su objetivo es determinar las acciones necesarias para garantizar y
documentar que el sistema se mantiene en un estado validado.
Revalidación
Repetición de la validación del proceso para garantizar
que los cambios en el proceso o equipo introducidos de conformidad con los
procedimientos de control de cambios no afectan negativamente a las
características del proceso ni a la calidad del producto.
Simulación de producto
Material cuyas características físicas y, cuando
corresponda, químicas (p. ej., viscosidad, tamaño de partículas, pH, etc.) son
muy parecidas a las del producto que se desea validar. En muchos casos, estas
características las cumple un lote de producto placebo.
Sistema
Conjunto de equipos con una finalidad común.
Validación concurrente
Validación efectuada durante la producción sistemática de
productos destinados a la venta.
Validación de la limpieza
La validación de la limpieza es la prueba documentada de
que un procedimiento de limpieza aprobado proporcionará equipos adecuados para
la elaboración de medicamentos.
Validación del proceso
Verificación documentada de que el proceso, realizado en
los parámetros establecidos, puede ofrecer resultados eficaces y reproducibles
para elaborar un medicamento que cumpla sus especificaciones y atributos de
calidad predeterminados.
Validación prospectiva
Validación llevada a cabo antes de la elaboración
sistemática de productos destinados a la venta.
Validación retrospectiva
Validación de un proceso para un producto que ya se ha
comercializado, basada en datos acumulados de fabricación, ensayos y lotes de
control.
ANEXO III
Liberación Paramétrica
1. PRINCIPIO
Es un sistema de liberación que ofrece la garantía de que
el producto es de la calidad deseada basándose en la información recogida
durante el proceso de fabricación y en el cumplimiento de exigencias
específicas de las Buenas Prácticas de Fabricación.
Para practicar la liberación paramétrica se debe cumplir
con las exigencias básicas de las Buenas Prácticas de Fabricación, un sistema
de Análisis de Peligro y Puntos Críticos de Control (HACCP) implementado, los
anexos aplicables y las directrices que se incluyen a continuación; y los
laboratorios deben ser autorizados por la ANMAT para tal fin.
2. LIBERACION PARAMETRICA
2.1 Se acepta que un conjunto completo de ensayos y
controles durante el proceso podría garantizar que el producto terminado cumple
las especificaciones.
2.2 La liberación paramétrica puede ser autorizada para
determinados parámetros específicos como alternativa a los ensayos rutinarios
de los productos terminados.
La autorización de la liberación paramétrica debe ser
concedida, denegada o retirada por la Autoridad Sanitaria Nacional. para cada producto o lote.
2.3 La liberación paramétrica se debe practicar en
presencia, supervisión y aprobación de una comisión de inspectores de las
Buenas Prácticas de Fabricación.
3. LIBERACION PARAMETRICA PARA PRODUCTOS ESTERILES
3.1 Esta sección solamente trata la parte de la liberación
paramétrica relacionada con la aprobación rutinaria de productos terminados sin
llevar a cabo un ensayo de esterilidad. La eliminación del ensayo de
esterilidad solamente será válida si se demuestra satisfactoriamente que se han
alcanzado las condiciones predeterminadas y validadas de esterilización.
3.2 El laboratorio debe contar con un sistema de garantía
de la esterilidad según se detalla en el glosario del presente anexo.
3.3 La liberación paramétrica se podrá autorizar si los
datos que demuestran la correcta elaboración del lote aportan, por sí solos,
garantía suficiente de que el proceso fue diseñado y validado para garantizar
la esterilidad del producto.
3.4 La liberación paramétrica solamente es aceptable para
productos esterilizados mediante esterilización terminal por calor húmedo, en
su envase final.
3.5 El fabricante debe contar con un historial de
cumplimiento adecuado de las normas de Buenas Prácticas de Fabricación.
3.6 Al evaluar el cumplimiento de las Buenas Prácticas de
Fabricación, deben tenerse en cuenta el historial de no esterilidad de los productos
y los resultados de los ensayos de esterilidad, llevados a cabo sobre el
producto en cuestión, junto con los productos procesados mediante el mismo
sistema de garantía de la esterilidad.
3.7 Es poco probable que un producto completamente nuevo
se considere adecuado para la liberación paramétrica, ya que los criterios de
aceptación incluirán un período de resultados satisfactorios del ensayos de
esterilidad. Puede darse el caso de que un producto nuevo no sea más que una
variación menor, desde el punto de vista de garantía de la esterilidad, y que
se puedan considerar aplicables por tanto, los datos existentes sobre el ensayo
de esterilidad de otros productos.
3.8 El diseño y la validación original del producto deben
garantizar que se mantiene su integridad en todas las condiciones operativas.
3.9 El sistema de control de cambios debe exigir la
revisión de los cambios por parte del personal de garantía de la esterilidad.
3.10 Debe estar implementado un sistema de Análisis de
Peligro y Puntos Críticos de Control (HACCP) en el sistema de garantía de la
esterilidad centrado en una evaluación de la aprobación como si se tratara de
productos no esterilizados.
3.11 La limpieza y sanitización de las partes de los
equipos, utensilios y todo el material que esté contacto directo con el
producto deben estar validadas.
3.12 Las áreas de elaboración del producto deben estar
calificadas como indica la legislación vigente para la exigencia del producto.
3.13 En el lugar de la elaboración y de la esterilización
deben estar presentes un profesional calificado en microbiología con
experiencia en la garantía de la esterilidad y la persona autorizada.
3.14 Debe existir un sistema para controlar la posible
contaminación microbiológica en el producto antes de la esterilización.
3.15 No debe existir la posibilidad de que se mezclen
productos esterilizados con productos no esterilizados, lo cual se debe
garantizar mediante barreras físicas o sistemas electrónicos validados.
3.16 Se debe verificar que los protocolos de esterilización
cumplan con las especificaciones mediante dos sistemas independientes, como
mínimo. Estos sistemas pueden ser dos personas o un sistema informático
validado más una persona.
3.17 Antes de la aprobación de cada lote de producto se
deben confirmar los siguientes puntos adicionales:
- En el equipo de esterilización utilizado se han
realizado todas las tareas de mantenimiento planificadas y las verificaciones
rutinarias.
- Todas las reparaciones y modificaciones han sido
aprobadas por el responsable de garantía de la esterilidad y la persona
autorizada.
- Todo el instrumental debe estar calibrado.
- El procedimiento de esterilización posee una validación
actualizada para la carga de producto procesado.
- La descripción del proceso de esterilización incluyendo
el tipo de ciclo, plantilla de registro, especificaciones de los parámetros del
ciclo (tiempo, temperatura, presión, F0) e indicadores químicos.
- Las especificaciones y métodos o procedimientos
aplicados para los controles en proceso. Ej: Pre-esterilización, carga
microbiana inicial, monitoreo de los parámetros del ciclo de esterilización y
verificación del registro de esterilización.
3.18 Una vez concedida la liberación paramétrica, las
decisiones sobre aprobación o rechazo de un lote se basarán en las
especificaciones aprobadas. El incumplimiento de las especificaciones para la
liberación paramétrica no podrá compensarse por haber superado un ensayo de
esterilidad.
GLOSARIO
Liberación paramétrica
Un sistema de aprobación que ofrece la garantía de que el
producto es de la calidad deseada basándose en la información recogida durante
el proceso de fabricación y en el cumplimiento de exigencias específicas de las
Buenas Prácticas de Fabricación.
Persona autorizada
Persona reconocida por la Autoridad Sanitaria Nacional, como Director Técnico y/o Co-Director Técnico del laboratorio
titular del registro, que tiene la responsabilidad de asegurar que cada lote
del producto terminado ha sido fabricado, analizado y aprobado para su liberación.
Sistema de garantía de la esterilidad
Suma de todas las medidas adoptadas para garantizar la
esterilidad de los productos. Para los productos esterilizados en forma
terminal, debe incluir las siguientes etapas:
(a) Diseño del producto.
(b) Conocimiento y control de la situación microbiológica
del material de partida y los componentes auxiliares de elaboración (como gases
y lubricantes).
(c) Control de la contaminación durante el proceso de
fabricación para evitar la entrada de microorganismos y su proliferación en el
medicamento.
(d) Prevención de mezclas entre los productos estériles y
no estériles.
(e) Mantenimiento de la integridad de los medicamentos.
(f) Proceso de esterilización.
(g) La totalidad del sistema de calidad en el que está
incluido el Sistema de garantía de la esterilidad, por ejemplo, control de
cambios, formación, procedimientos escritos, verificaciones de la aprobación,
mantenimiento preventivo planificado, análisis de modo de fallos, sistema de
Análisis de Peligro y Puntos Críticos de Control, prevención de errores
humanos, validación, calibración, etc.
ANEXO IV
ESTANDARES
PARA ENSAYOS FISICO - QUIMICOS
1- CONSIDERACIONES GENERALES
Las Sustancias de Referencia y los Estándares necesarios
para la implementación de los diversos métodos analíticos que figuran en las
monografías, contribuyen a resguardar la calidad de los resultados obtenidos.
La ausencia de dichas referencias de materias primas codificadas o no
codificadas, sustancias relacionadas e impurezas, dificulta las posibilidades
analíticas correspondientes. La preparación de Estándares Secundarios y
Estándares de Trabajo por parte de los usuarios, tiende a resolver la carencia
de los mencionados materiales, haciéndose necesario en estos casos, establecer
criterios que permitan garantizar la confiabilidad de los mismos. De igual
manera contribuirán adecuados lineamientos que abarquen el acondicionamiento,
fraccionamiento, conservación, manipuleo y reanálisis de dichos materiales.
2- ESTANDARES SECUNDARIOS DE MATERIAS PRIMAS Y DE TRABAJO
DE: MATERIAS PRIMAS
2.1-De sustancias codificadas en "Farmacopea
Argentina" y/o en alguna de las "Farmacopeas Internacionalmente
Reconocidas". (Estándares Secundarios)
2.1.1.- ORIGEN DE LA MATERIA PRIMA
La materia prima tendrá los siguientes orígenes
alternativos:
a.- Elaboradores o proveedores de la sustancia
b.- Síntesis propia realizada en el país
c.- Lotes de materia prima purificada, adquiridos para la
elaboración de productos
e.- Lotes de materia prima tal cual, adquiridos para la
elaboración de productos
2.1.2.- ANALISIS A REALIZAR.
Estos "Estándares Secundarios" serán traceables
a una "Sustancia de Referencia" proveniente de la "Farmacopea
Argentina" o de alguna de las "Farmacopeas Internacionalmente
Reconocidas"
Los análisis a realizar sobre la materia prima serán los
incluidos en la monografía de la Farmacopea de la cual provenga la Sustancia de Referencia a utilizar.
Se realizarán no menos de cuatro ensayos / determinaciones
independientes de cada uno de los métodos cuyos resultados sean cuantificables
(punto de fusión, contenido de agua o pérdida por secado, absortividad
específica, impurezas comunes e impurezas quirales, poder rotatorio, valoración
volumétrica, valoración por cromatografía líquida de alta performance, debiendo
aumentarse las mismas cuando las dispersiones obtenidas no sean satisfactorias.
Este criterio incluye la determinación del factor de la solución titulante en
las volumetrías.
2.2- De sustancias no codificadas en "Farmacopea
Argentina" ni en ninguna de las "Farmacopeas Internacionalmente
Reconocidas" ("Estándares de Trabajo")
2.2.1.- ORIGEN DE LA MATERIA PRIMA
Tendrán los siguientes orígenes alternativos:
a) Casa matriz del propio laboratorio innovador de la
sustancia
b) Elaboradores no innovadores o proveedores de estándares
de la sustancia.
c) Síntesis propia realizada en el país.
d)Lotes de materia prima purificada adquiridos para la
elaboración de productos.
e) Lotes de materia prima tal cual, adquiridos para la elaboración
de productos.
2.2.2.- ANALISIS A REALIZAR
A los fines de garantizar la validez de la identificación,
la valoración y la pureza de las sustancias involucradas, surge la necesidad de
reforzar con algunas técnicas no tradicionales el análisis de las mismas,
generalizándose el siguiente listado de metodologías:
• Espectrofotometría infrarroja.
• Caracterización estructural por Resonancia Magnética
Nuclear (RMN) y Espectroscopía de masas (EM).
• Determinación del Punto de fusión (PF).
• Calorimetría diferencial de barrido (CDB).
• Determinación del contenido de agua (por Karl Fischer o
Coulombimetría) y/o Pérdida por desecación.
• Espectrofotometría ultravioleta.
• Polarimetría.
• Cromatografía en capa delgada (CCD).
• Cromatografía líquida de alta eficacia (CLAE), con
detector de arreglo de diodos.
• Volumetría.
La espectrofotometría infrarroja debe realizarse de manera
de no alterar el estado sólido de la muestra.
La caracterización estructural, conducente a determinar la
identidad de la sustancia, se realizará a través de técnicas que permitan,
según el caso, la correcta asignación de los átomos de C y de H constitutivos
de la molécula, como por ejemplo a través de resonancia magnética nuclear
asociada con espectroscopía de masas. De esta manera deberán descartarse
isómeros y/o sustancias de estructuras estrechamente relacionadas. La
mencionada caracterización será requerida solamente sobre el primer estándar de
la sustancia, proveniente de alguno de los puntos 2.1.b al 2.1.d, que el
usuario haya adecuado a los requerimientos de "Farmacopea Argentina".
Para los posteriores estándares de la misma materia prima, el conjunto de los
análisis detallados en la TABLA I, con prescindencia de la caracterización
estructural, permitirá abarcar los requerimientos cualitativos y cuantitativos
correspondientes.
El punto de fusión no se determinará cuando la
descomposición producida por el calentamiento interfiera con el ensayo.
El perfil calorimétrico de las sustancias a través de
calorimetría diferencial de barrido, contribuye significativamente tanto a
identificar las mismas, como a evaluar el estado sólido en relación a las
formas amorfa o polimórficas presentes.
Si la molécula de la sustancia posee centros, planos o
ejes quirales, es necesario evaluar si la misma se presenta en forma de mezclas
racémicas o como alguno de los estereoisómeros puro o impurificado. La
determinación del poder rotatorio provee información al respecto. Se obtienen
resultados significativos con adecuados sistemas quirales de cromatografía
líquida de alta eficacia, que demandan menor cantidad de muestra y aportan
información más específica sobre el tema.
Las impurezas desconocidas determinadas por cromatografía
en capa delgada o por cromatografía líquida de alta eficacia no deben exceder,
individualmente, el 0,2 % de la intensidad o del área de la sustancia
principal, respectivamente, y su sumatoria no debe exceder el 0,5 % de los
mismos valores.
Los métodos isocráticos de cromatografía líquida de alta
eficacia a ensayar deben complementarse, durante su desarrollo, con la
aplicación de gradientes antes de finalizar las corridas, para detectar la
presencia de la mayor cantidad posible de impurezas presentes, y con el estudio
de "pureza de picos", a través de un "detector con arreglo de
diodos".
Será necesario contar, en lo posible, con testigos de
impurezas y de sustancias relacionadas, especialmente de aquellas consideradas
tóxicas.
Una vez conocidos los alcances de los métodos
cromatográficos estudiados, se seleccionará el más eficiente para incorporarlo
a la rutina de control subsiguiente, en tanto sus resultados no sean
complementarios con los de otro método, en cuyo caso deberán utilizarse ambos.
Si la valoración volumétrica no fuera específica, las
impurezas y las sustancias relacionadas deberán ser detectables por
cromatografía en capa delgada o por cromatografía líquida de alta eficacia,
excepto cuando tengan actividad farmacológica similar a la del principio
activo.
Los métodos enunciados pueden ser reemplazados por otros
equivalentes o de aún mayor especificidad, en tanto éstos cumplan con el
objetivo del análisis al cual sustituyen.
Los métodos presentados deberán estar validados por el
usuario o por la bibliografía que los avale, la cual deberá ser de nivel reconocido.
TABLA I
ANALISIS A REALIZAR SOBRE MATERIAS PRIMAS
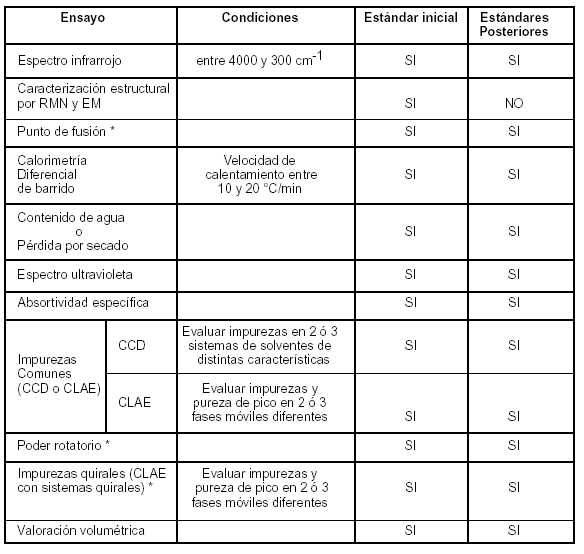
* Análisis a realizar cuando corresponda.
3.-ESTANDARES DE SUSTANCIAS RELACIONADAS E IMPUREZAS
DEFINIDAS POR LA AUTORIDAD SANITARIA NACIONAL
3.1.- ORIGEN.
Las "Sustancias de Referencia" y los
"Estándares", provendrán de las siguientes alternativas:
• "Farmacopea Argentina" y/o "Farmacopeas
Internacionalmente Reconocidas".
• Elaboradores o proveedores
• Casa matriz del laboratorio.
• Síntesis local propia.
3.2.- ANALISIS A REALIZAR
Sólo corresponde confirmar estructuralmente la identidad
de los "estándares" provenientes de:
• Elaboradores o proveedores
• Síntesis local propia
4.-ACONDICIONAMIENTO, FRACCIONAMIENTO, CONSERVACION,
MANIPULEO Y REANALISIS
Lineamientos generales
• Se conservarán a temperatura y humedad adecuadas
(generalmente a 5 ? 1 ºC) y protegidos de la luz.
• Durante el manipuleo deben tomarse las siguientes
precauciones:
a) Abrir el envase cuando haya alcanzado temperatura
ambiente.
b) Proteger de la luz si la sustancia es fotosensible.
c) Proteger de la humedad durante la pesada si la
sustancia es higroscópica.
• Las muestras seleccionadas como potenciales estándares
deben homogeneizarse adecuadamente antes de comenzar los ensayos sobre las
mismas.
• Una vez finalizados los ensayos, si los resultados son
satisfactorios, las muestras deben fraccionarse en cantidades convenientes,
preferentemente no mayores a un gramo, empleando envases de vidrio inactínico,
u otro material apropiado, con cierre hermético.
• En el rótulo de los envases debe constar:
a) Nombre de la sustancia.
b) Números de lote y de identificación del envase.
c) Título (sobre sustancia tal cual, anhidra o desecada).
d) Fecha de reanálisis.
e) Fecha de vencimiento (si correspondiera).
f) Número de veces que fue abierto el envase.
• Reanalizar la sustancia en función de la estabilidad de
la misma (cada 2 ó 3 años, o con mayor frecuencia en casos de sustancias
altamente inestables)
• Reemplazar la sustancia cuando se detecte o se presuma
degradación significativa
GLOSARIO
Sustancia de Referencia Farmacopea Argentina:
Material de uniformidad comprobada, cuya monografía ha
sido incluida en la "Farmacopea Argentina", desarrollado total o
parcialmente por A.N.M.A.T. - I.NA.ME y avalado por dicha Farmacopea, cuyo
empleo se reserva a ensayos químicos y físicos específicos en los que se comparan
sus propiedades con las de un producto problema y que posee un grado de pureza
adecuado para el uso al que se destina.
Sustancia de Referencia Farmacopea Argentina Certificada:
Material de uniformidad comprobada, cuya monografía ha
sido incluida en la "Farmacopea Argentina", desarrollado a través de
ensayos colaborativos avalados por dicha Farmacopea y A.N.M.A.T. - I.NA.ME,
cuyo empleo se reserva a ensayos químicos y físicos específicos en los que se
comparan sus propiedades con las de un producto problema y que posee un grado
de pureza adecuado para el uso al que se destina.
Estándar Secundario:
Material de uniformidad comprobada, cuya monografía ha
sido incluida en la "Farmacopea Argentina" y/o en alguna de las
"Farmacopeas Internacionalmente Reconocidas", desarrollado localmente
por el usuario, cuyo empleo se reserva a ensayos químicos y físicos específicos
en los que se comparan sus propiedades con las de un producto problema y que
posee un grado de pureza adecuado para el uso al que se destina.
Estándar de Trabajo:
Material de uniformidad comprobada, cuya monografía no ha
sido incluida en la "Farmacopea Argentina" ni en ninguna de las
"Farmacopeas Internacionalmente Reconocidas", desarrollado localmente
por el usuario, cuyo empleo se reserva a ensayos químicos y físicos
específicos, en los que se comparan sus propiedades con las de un producto
problema y que posee un grado de pureza adecuado para el uso al que se destina.
ANEXO V
Calificación de Proveedores de material de
acondicionamiento para la industria farmacéutica
1. INTRODUCCION
1.1 Principio: La
industria farmacéutica debe asegurar que los materiales de acondicionamiento
adquiridos cumplan con los requisitos de calidad especificados. Una de las
herramientas para asegurarlo es auditar y calificar a los proveedores de los
mismos.
1.2 El objetivo del presente anexo es establecer pautas
básicas para la auditoría y calificación de proveedores de materiales de
acondicionamiento, con el fin garantizar la calidad de sus provisiones.
1.3 Todo el proceso de auditoría y calificación deberá
estar orientado a cumplir con los siguientes objetivos:
a) Disponer de materiales que reúnan las especificaciones
establecidas,
b) Disminuir el rechazo de materiales no conformes
c) Mejorar la confiabilidad de los sistemas de calidad de
los proveedores.
2. REQUISITOS PREVIOS
2.1 El jefe del Departamento de Garantía de Calidad es la
persona responsable de realizar o designar un responsable para realizar las
auditorías y calificaciones de los proveedores de materiales de
acondicionamiento.
2.2 El Jefe / Responsable del departamento de Garantía de la Calidad del titular del registro del Registro podrá tercerizar la ejecución de la auditoría
de un proveedor en una institución, sin fines de lucro, dedicada a tal fin (preferentemente
con especialidad en calidad) que no tenga conflictos de intereses entre las
partes. La institución deberá contar con la Autorización de la Autoridad Sanitaria Nacional.
2.3 El responsable de auditar y calificar un proveedor de
materiales de acondicionamiento deberá contar, como mínimo, con:
a- Un programa de concientización de criterios y conceptos
de calidad para los proveedores.
b- Información histórica de los proveedores
c- Procedimientos operativos normatizados que incluyan:
• programas de auditorías
• plan de auditoría
• guías y/o cuestionarios de verificación para la
auditoría
• formato para el informe de la auditoría
• metodología para la evaluación y calificación de
proveedores
3. PROCEDIMIENTO
3.1- Las auditorías se deben planificar y programar según
procedimientos operativos normatizados.
3.2- Los procedimientos operativos normatizados deben
contar con guías y/o cuestionarios de auditorías. Con el fin de unificar los
criterios de calificación, los lineamientos en que se basan estas guías y/o
cuestionarios deben ser previamente consensuados con la Autoridad Sanitaria Nacional.
3.3 Deberá establecerse una metodología para la
clasificación de proveedores en función de los resultados de las auditorías y
su calificación. Los parámetros de esta clasificación estarán consensuados con la Autoridad Sanitaria Nacional.
3.4- El Departamento de Garantía de Calidad como
responsable de la auditoría y calificación de proveedores deberá disponer de un
registro de Proveedores calificados con su clasificación.
3.5- Todas las auditorías y calificaciones deben estar
documentadas y registradas.
3.6- La Autoridad Sanitaria Nacional tendrá acceso a registros, resultados y calificación de los proveedores auditados.
3.7- El plan de auditoría deberá establecer la frecuencia
de las auditorías de acuerdo con la clasificación del proveedor o según lo
definido previamente por el Departamento de Garantía de Calidad.
3.8- Algunas de las herramientas recomendadas para
modificar la calificación y clasificación de un proveedor son:
• Verificación de la implementación de un plan de acciones
correctivas y preventivas por parte del proveedor
• Mayor frecuencia en las auditorías
• Muestreos y niveles de inspección especiales.
ANEXO VI
Buenas Prácticas de Fabricación para Ingredientes
Farmacéuticos Activos
1 INTRODUCCION
1.1 Objetivo
Este documento está pensado para proveer lineamientos
generales para la aplicación de la Buenas Prácticas de Fabricación (BPF) para la manufactura de ingredientes farmacéuticos activos (APIs) bajo un sistema
apropiado de manejo de la calidad. Se intenta asegurar que los APIs satisfagan
los requerimientos de calidad y pureza que implican o dicen tener.
En este Anexo "Fabricación" incluye todas las
operaciones de recepción de materiales, producción, envasado, reenvasado,
etiquetado, reetiquetado, control de calidad, liberación, almacenamiento y
distribución de APIs y los controles relacionados.
El presente documento, como un todo no cubre aspectos de
seguridad para el personal comprometido en la manufactura ni aspectos de
protección para el medio ambiente. Estos controles son responsabilidad
inherente al elaborador y están regidos por normativas específicas.
1.2 Aplicación Regulatoria
Dentro de la comunidad mundial, los materiales pueden
variar en su clasificación legal como API. Cuando un material es clasificado
como un API en la región o país en el cual es elaborado o usado en un
medicamento, debe elaborarse acorde a este documento.
1.3 Alcances
Se aplica a la elaboración de APIs para uso en medicina
humana (productos medicinales de uso humano).
Este anexo abarca APIs elaborados por síntesis química,
extracción, fermentación/cultivo de células, recolección de fuentes naturales o
alguna combinación de estos procesos.
Un "Material de Partida API" es un material de
partida, intermediario o API que es usado en la producción de otro API, y que
es incorporado como un fragmento estructural significativo en la estructura del
mismo. Puede ser un artículo comercial, un material comprado de uno o más
proveedores bajo contrato de acuerdo comercial o producido en el laboratorio.
Un material de partida API normalmente tiene definida la estructura y las
propiedades químicas.
La compañía debe señalar y documentar los fundamentos del
punto en el cual la producción del API comienza. Para procesos de síntesis, es
conocido como el punto en el cual el "Material de Partida API" es
introducido en el proceso. Para otros procesos (por ejemplo fermentación,
extracción, purificación, etc.) el fundamento deberá establecerse caso por
caso. La tabla I da una guía del punto en el cual el "Material de Partida
API" es introducido normalmente en el proceso.
A partir de este punto, deben aplicarse a este
intermediario y/o etapas de elaboración del API las BPF apropiadas - como se
define en esta guía -. Esto incluye la validación de las etapas críticas del
proceso determinando el impacto en la calidad del API. Sin embargo, debe
hacerse notar que el hecho que una compañía elija validar una etapa del proceso
no necesariamente define a esa etapa como crítica.
Los requerimientos BPF en este documento deben aplicarse
en las etapas grisadas y con letra italica en la tabla I. Esto no implica que
todas las etapas descriptas en la tabla deban completarse. La rigurosidad de
BPF en la elaboración de APIs debe incrementarse a medida que el proceso avanza
desde las primeras etapas a las finales, purificación y embalaje. Los procesos
físicos de APIs tales como granulación, recubrimiento o manipulación física de
tamaño de partícula (por ejemplo micronizado, molienda) deben manejarse al
menos con los estándares de esta guía.
Este anexo de BPF no se aplica a etapas previas a la
introducción del definido "Material de Partida API".
Tabla I: Aplicación de esta guía a la elaboración de APIs
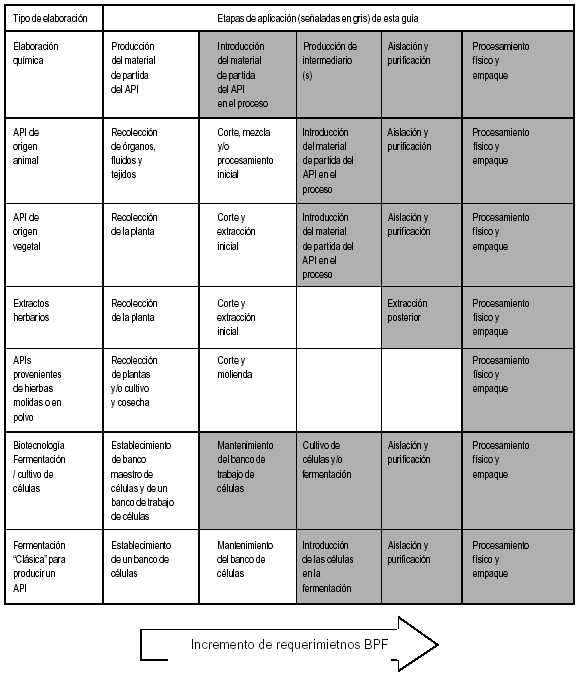
2 GERENCIA DE CALIDAD
Principios
2.1 La calidad debe ser responsabilidad de todas las
personas involucradas en la manufactura.
2.2 Cada elaborador debe establecer, documentar e
implementar un sistema efectivo para procurar calidad que involucre la
participación activa de la gerencia y el personal de manufactura apropiado.
2.3 El sistema de manejo de calidad debe abarcar la
estructura de organización, procedimientos, procesos y recursos, así como
actividades necesarias para asegurar confidencialidad, para que los APIs
satisfagan las especificaciones propuestas de calidad y pureza. Todas las
actividades relacionadas con la calidad deben estar definidas y documentadas.
2.4 Debe existir una/s unidad/es de calidad independiente
de producción y que satisfaga las responsabilidades de aseguramiento de la
calidad (QA) así como las responsabilidades de control de calidad (QC). Pueden
presentarse como unidades separadas de QA y QC o como una única unidad,
dependiendo del tamaño y estructura de la organización.
2.5 Deben especificarse las personas autorizadas para
liberar intermediarios y APIs.
2.6 Todas las actividades relacionadas con calidad deben
registrarse en el momento en que se realizan.
2.7 Cualquier desviación de los procedimientos
establecidos debe documentarse y explicarse. Desviaciones críticas deben ser
investigadas y la investigación y sus conclusiones deben documentarse.
2.8 Ningún material debe liberarse o usarse antes de
completar satisfactoriamente la evaluación por las unidades de calidad, a menos
que en el lugar haya sistemas apropiados que permitan su uso (por ejemplo,
liberar en cuarentena como se describe en la sección "Procedimientos de
distribución", o el uso de materiales de partida o intermediarios
pendientes de evaluación completa).
2.9 Deben existir procedimientos para notificar
oportunamente a la gerencia responsable de inspecciones regulatorias, serias
deficiencias BPF, productos con defectos y acciones relacionadas (por ejemplo,
quejas relacionadas con calidad, recolección de mercado, acciones regulatorias,
etc.).
Responsabilidades de Unidad(es) de Calidad
2.10 La(s) unidad(es) de calidad debe involucrarse en
todas las materias relacionadas con la calidad.
2.11 La(s) unidad(es) de calidad debe revisar y aprobar
todos los documentos apropiados relacionados con calidad.
2.12 Las responsabilidades de mayor importancia de la(s)
unidad(es) de calidad independiente no deben delegarse. Estas responsabilidades
deben estar descriptas por escrito y deben incluir, pero no necesariamente
estar limitadas a:
1. Liberación o rechazo de todos los APIs. Liberación o
rechazo de intermediarios para uso fuera de la compañía elaboradora;
2. Establecer un sistema de liberación o rechazo de
materiales de partida, intermediarios y materiales de acondicionamiento y
rotulado;
3. Revisión completa de los registros del lote de
producción y de los controles del laboratorio de las etapas críticas del
proceso antes de la liberación del API para su distribución;
4. Asegurar que las desviaciones críticas sean
investigadas y resueltas;
5. Aprobar todas las especificaciones e instrucciones de la Orden Maestra de Producción;
6. Aprobar todos los procedimientos que impactan la
calidad de intermediarios o APIs;
7. Asegurar que se realicen auditorías internas
(autoinspecciones);
8. Aprobar intermediarios o APIs elaborados por contrato;
9. Aprobar cambios que potencialmente impacten en la
calidad de intermediarios o APIs;
10. Revisar y aprobar protocolos de validación e informes;
11. Asegurar que las quejas relacionadas con la calidad
son investigadas y resueltas;
12. Asegurar que existen sistemas efectivos para el
mantenimiento y calibración del equipamiento crítico;
13. Asegurar que los materiales son testeados
apropiadamente y los resultados son informados;
14. Asegurar que los datos de estabilidad permitan
revaloraciones, fijar fechas de vencimiento y condiciones de almacenaje en APIs
y/o intermediarios, cuando corresponda;
15. Realizar revisiones de calidad del producto (como se
define en "Revisión de calidad del producto").
Responsabilidades para las Actividades de Producción
2.13 La responsabilidad de las actividades de producción
deben describirse por escrito y deben incluir, pero no necesariamente limitarse
a:
1. Preparación, revisión, aprobación y distribución de
instrucciones para la producción de intermediarios o APIs acorde a procedimientos
escritos;
2. Producción de APIs, y cuando corresponda,
intermediarios acorde a instrucciones preaprobadas;
3. Revisión de todos los registros de lote de producción y
asegurarse que estén completos y firmados;
4. Asegurar que todas las desviaciones de producción son
comunicadas, evaluadas, y que las desviaciones críticas son investigadas y las
conclusiones registradas;
5. Asegurar que las instalaciones de producción se
encuentren limpias y con adecuada desinfección;
6. Asegurar que las calibraciones necesarias hayan sido
realizadas y se mantengan los registros;
7. Asegurar que el equipamiento y sus instrucciones de uso
sean mantenidos y los registros sean llevados;
8. Asegurar que los protocolos de validación y los registros
son revisados y aprobados;
9. Evaluación de las propuestas de cambio en el producto,
proceso o equipamiento;
10. Asegurar que los cambios en las instalaciones y
equipamiento sean calificados, cuando corresponda.
Auditorías Internas (Autoinspecciones)
2.14 Para cumplir con los principios de BPF para APIs
deben realizarse auditorías internas regulares de acuerdo con un programa
aprobado.
2.15 Los resultados de las auditorías y las acciones
correctivas deben documentarse y ser consideradas por el responsable de la
firma. Las acciones correctivas convenidas deben completarse en tiempo y forma.
Revisión de Calidad del Producto
2.16 Deben efectuarse revisiones periódicas de calidad de
APIs con el objetivo de verificar la consistencia del proceso. Normalmente,
tales revisiones deben ser realizadas y documentadas en forma anual y deben
incluir al menos:
- Una revisión de resultados de controles críticos en
proceso y de ensayos críticos en APIs;
- Una revisión de todos los lotes que no cumplieron con las
especificaciones establecidas;
- Una revisión de todos los desvíos críticos o de no
conformidad e investigaciones relacionadas;
- Una revisión de todo cambio realizado en el proceso o
método analítico;
- Una revisión de los resultados del monitoreo del
programa de estabilidad;
- Una revisión de todos los rechazos relacionados con la
calidad, quejas y recolección del mercado;
- Una revisión de adecuación de acciones correctivas.
2.17 Los resultados de estas revisiones deben evaluarse y
debe emprenderse una estimación de acciones correctivas o de revalidación. Las
razones para tales acciones correctivas deben documentarse. Las acciones
correctivas convenidas deben completarse en tiempo y forma.
3 PERSONAL
Calificación de Personal
3.1 Debe haber un número adecuado de personal calificado
con la apropiada educación, experiencia y/o entrenamiento para llevar a cabo y
supervisar la elaboración de intermediarios y APIs.
3.2 Las responsabilidades de todo el personal involucrado
en la elaboración de intermediarios y APIs deben estar especificadas por
escrito.
3.3 El entrenamiento debe hacerse regularmente por
personas calificadas y debe cubrir como mínimo las operaciones que cada
empleado realiza y las GMP relacionadas con las funciones del empleado. Deben
mantenerse registros de entrenamiento y estos deben ser periódicamente
evaluados.
Higiene del Personal
3.4 El personal debe cumplir con hábitos de salud e
higiene.
3.5 El personal debe vestir ropa limpia y adecuada para la
actividad de elaboración con la cual está involucrado, y debe cambiarse cuando
corresponda. La ropa protectora adicional, para cabeza, cara, manos y brazos
debe usarse cuando sea necesario, para proteger intermediarios y APIs de la
contaminación.
3.6 El personal debe evitar el contacto directo con
intermediarios o APIs.
3.7 Fumar, comer, beber, mascar, y el almacenamiento de
alimentos, debe restringirse a áreas asignadas separadas de las de elaboración.
3.8 El personal que sufre enfermedades infecciosas o con lesiones
abiertas en superficies expuestas del cuerpo no debe ocuparse en actividades
que puedan comprometer la calidad de los APIs. Cualquier persona que muestre en
algún momento (tanto por examen médico o por observación de un supervisor)
tener una aparente enfermedad o lesión abierta, debe excluirse de las
actividades en las cuales las condiciones de salud puedan afectar adversamente
la calidad del APIs, hasta que las condiciones sean corregidas o el personal
médico calificado determine que la inclusión de la persona no arriesga la
seguridad o calidad del APIs.
Consultores
3.9 Los consultores asesores de manufactura y control de
intermediarios y APIs deben tener suficiente educación, entrenamiento y
experiencia o una combinación de todo ello, para asesorar en la materia para la
cual son contratados.
3.10 Deben llevarse registros exponiendo nombre, dirección
y calificaciones del consultor, y tipo de servicio provisto por éste.
4 EDIFICIOS E INSTALACIONES (SERVICIOS DE SOPORTE)
Diseño y Construcción
4.1 Los edificios e instalaciones usados en la manufactura
de intermediarios y APIs deben ubicarse, diseñarse y construirse facilitando la
limpieza, el mantenimiento y las operaciones apropiadas al tipo y etapa de
elaboración. Las instalaciones deben diseñarse para minimizar la potencial
contaminación. Si se hubiesen establecido especificaciones microbiológicas para
intermediarios o APIs, las instalaciones deben diseñarse en forma apropiada
para limitar la exposición a contaminaciones microbiológicas inconvenientes.
4.2 Los edificios e instalaciones deben tener espacio
adecuado para la ubicación ordenada del equipamiento y mantenerlo para prevenir
confusiones y contaminación.
4.3 Todo equipamiento que posea por sí mismo protección
adecuada del material (por ejemplo, sistema cerrado) puede ubicarse en el
exterior.
4.4 El flujo de materiales y personal a través del
edificio o anexos debe diseñarse para prevenir confusiones o contaminación.
4.5 Deben definirse áreas u otros sistemas de control para
las siguientes actividades:
- Recepción, identificación, muestreo y cuarentena de
materiales entrantes, pendientes de liberación;
- Cuarentena antes de la liberación de intermediarios y
APIs;
- Muestreo de intermediarios y APIs;
- Permanencia de materiales rechazados antes de la
disposición final (por ejemplo, devolución, reprocesado o destrucción);
- Almacenamiento de materiales liberados;
- Operaciones de producción;
- Acondicionamiento y etiquetado;
- Operaciones de laboratorio.
4.6 Debe proveerse al personal de los sanitarios
adecuados, provistos de agua fría y caliente, así como de jabón o detergente
apropiados, secadores de aire o servicio de toallas individuales. Deben estar
separados de áreas de elaboración, pero con fácil acceso desde las mismas.
Cuando sea necesario debe proveerse de adecuadas áreas de duchas o cambio de
ropa.
4.7 Las áreas de laboratorio deben estar separadas del
área de producción. Aquellas usadas para controles en proceso, pueden ubicarse
en áreas de producción, teniendo en cuenta que las operaciones del proceso de
producción no afecten adversamente la exactitud de las medidas de laboratorio,
y el laboratorio y sus operaciones no afecten los procesos de producción de
intermediarios o APIs.
Servicios
4.8 Todos los servicios que puedan afectar la calidad del
producto (por ejemplo: vapor, gases, aire comprimido y calefacción, ventilación
y acondicionamiento de aire) deben estar calificados y monitoreados
convenientemente y deberán tomarse acciones cuando los límites sean excedidos.
Debe disponerse de esquemas para estos sistemas de servicios.
4.9 Debe proveerse cuando sea necesario de adecuada
ventilación, filtración de aire y sistemas de extracción. Estos sistemas
deberán diseñarse y construirse minimizando riesgos de contaminación y
contaminación cruzada, y deberán incluir equipos para control de la presión de
aire, microorganismos (si corresponde), polvo, humedad y temperatura en forma
adecuada durante la etapa de elaboración. Se deberá prestar especial atención a
aquellas áreas donde los APIs se encuentren expuestos al medio ambiente.
4.10 En el caso que el aire sea recirculado a áreas de
producción, deben tomarse medidas apropiadas para el control de riesgo de
contaminación cruzada.
4.11 Todas las cañerías instaladas deben estar
apropiadamente identificadas. Además puede existir identificación de líneas
individuales, documentación, sistemas de control computarizado, o medios
alternativos. Las cañerías deben ubicarse apropiadamente para evitar el riesgo
de contaminación de intermediarios o APIs.
4.12 Los drenajes deben ser de tamaño adecuado y deben
estar provistos de una toma de aire o un dispositivo adecuado para evitar
reflujo, cuando corresponda.
Agua
4.13 El agua usada en la elaboración de APIs debe ser
adecuada para dicho uso.
4.14 El agua de proceso debe como mínimo seguir la guía de
OMS para calidad de agua potable, a menos que se justifique otra calidad.
4.15 Cuando el agua potable es insuficiente para asegurar
la calidad del API, y son exigidas estrictas especificaciones químicas y/o
microbiológicas de calidad de agua, deben establecerse especificaciones
apropiadas para parámetros físico/químicos, recuento microbiano total,
organismos objetables y/o endotoxinas.
4.16 Si el agua utilizada en el proceso es tratada por el
elaborador para alcanzar una determinada calidad, el proceso de tratamiento
debe validarse y monitorearse con límites apropiados de acción.
4.17 Cuando se elabora un API no estéril destinado a la
producción de un medicamento estéril, el agua utilizada en la aislación final y
en las etapas de purificación debe monitorearse y controlarse respecto del
recuento microbiano total, organismos objetables y endotoxinas.
Areas de Contención
4.18 En la producción de materiales sensibilizantes tales
como penicilinas y cefalosporinas, las áreas de producción deben ser dedicadas.
Pueden incluir instalaciones, unidades manejadoras de aire y/o equipos de
producción.
4.19 Areas de producción dedicadas deberán también
utilizarse cuando se trate de materiales de naturaleza infecciosa, de alta
actividad farmacológica o toxicidad (por ejemplo, ciertos esteroides o agentes
citotóxicos anticancerosos) a menos que los procedimientos de limpieza y/o
inactivación estén validados y se mantengan.
4.20 Deben establecerse e implementarse medidas apropiadas
para prevenir contaminación cruzada entre personal, materiales, etc., que se
trasladen de un área dedicada a otra.
4.21 Ninguna actividad productiva (incluyendo pesada,
molienda o envasado) de material no farmacéutico, altamente tóxico tales como
herbicidas y pesticidas, no deberá realizarse utilizando las instalaciones y/o
equipamiento usado para la producción de APIs. El manejo y almacenamiento de
estos materiales deberá estar separado de los APIs.
Iluminación
4.22 En todas las áreas debe proveerse de adecuada
iluminación para facilitar la limpieza, el mantenimiento y las distintas
operaciones.
Desagües y Residuos
4.23 Todos los residuos y desechos (por ejemplo, sólidos,
líquidos o gases provenientes de la elaboración) producidos en y desde los
edificios y las áreas adyacentes deben eliminarse oportunamente de manera
inocua y en forma sanitaria. Los contenedores y los conductos para materiales
de desecho deben estar claramente identificados.
Sanitización y Mantenimiento
4.24 Los edificios utilizados para la elaboración de
intermediarios y APIs deben estar mantenidos apropiadamente y en condiciones de
limpieza adecuadas.
4.25 Deben establecerse procedimientos escritos asignando
responsabilidades para la sanitización y describiendo programas, métodos y
equipamiento de limpieza y los materiales utilizados en los edificios e
instalaciones.
4.26 Cuando sea necesario, deben también ser establecidos
procedimientos escritos para el uso adecuado de rodenticidas, insecticidas,
fungicidas y agentes fumigantes de limpieza y sanitizantes, para prevenir la
contaminación del equipamiento, de los materiales de partida, de embalaje,
etiquetado, intermediarios y APIs.
5 EQUIPAMIENTO DE PROCESO
Diseño y Construcción
5.1 El equipamiento usado en la manufactura de
intermediarios y APIs debe ser de apropiado diseño y tamaño adecuado, y estar
correctamente ubicado para el uso previsto, su limpieza, sanitización (si
corresponde) y mantenimiento.
5.2 El equipamiento debe estar construido de forma tal que
las superficies en contacto con los materiales de partida, intermediarios o
APIs no alteren su calidad, más allá de especificaciones oficiales u otras.
5.3 El equipamiento de producción debe usarse sólo dentro
del rango de su calificación de operación.
5.4 El equipamiento principal (por ejemplo reactores,
contenedores de almacenamiento) y líneas de proceso instaladas de manera
permanente usados durante la producción del intermediario o API, deben estar
adecuadamente identificados.
5.5 Ninguna sustancia asociada con el funcionamiento del
equipamiento tales como lubricantes, fluidos de enfriamiento o calefacción,
deben estar en contacto con los intermediarios o APIs ni alterar su calidad,
más allá de especificaciones oficiales u otras. Cualquier desviación de esto
debería evaluarse para asegurar que no hay detrimento en la aptitud del
material. Deben usarse, cuando sea posible, lubricantes y aceites grado
alimenticio.
5.6 Cuando sea necesario debe usarse equipamiento cerrado
o contenido. Si se utiliza un equipo abierto o se procede a la apertura del
mismo, deben tomarse adecuadas precauciones para minimizar el riesgo de
contaminación.
5.7 Debe mantenerse un conjunto de esquemas actualizados
de equipamiento e instalaciones críticas (por ejemplo, instrumentación).
Mantenimiento y Limpieza del Equipamiento
5.8 Deben establecerse programas y procedimientos
(incluyendo asignación de responsabilidades) para el mantenimiento preventivo
del equipamiento.
5.9 Deben establecerse procedimientos escritos para la
limpieza del equipamiento y su subsecuente liberación de uso para la
elaboración de intermediarios y APIs. Los procedimientos de limpieza deben
contener suficientes detalles para permitir que operarios la efectúen en cada
equipo de manera efectiva y reproducible. Estos procedimientos deberán incluir:
- Asignación de responsabilidad para la limpieza del
equipamiento;
- Programas de limpieza incluyendo, cuando sea necesario,
programas de sanitización;
- Descripción completa de métodos y materiales incluyendo dilución
de agentes limpiadores utilizados;
- Cuando corresponda, instrucciones para desarmar y
rearmar cada parte del equipo para asegurar una adecuada limpieza;
- Instrucciones para remover o destruir la identificación
del lote anterior;
- Instrucciones para la protección de la contaminación del
equipo limpio, previo su uso;
- Inspección del equipo para la limpieza inmediatamente
antes del uso;
- Cuando corresponda, establecer el máximo tiempo que
puede transcurrir entre el momento en que se completa el proceso y la limpieza
del equipamiento.
5.10 Los equipos y utensilios deben limpiarse, guardarse y
cuando corresponda sanitizarse o esterilizarse para prevenir contaminación o
arrastre de material anterior que podría alterar la calidad del intermediario o
API, más allá de especificaciones oficiales u otras.
5.11 Cuando el equipamiento está asignado a producción
continua o campaña de sucesivos lotes del mismo intermediario o API, el
equipamiento debe limpiarse en intervalos apropiados para prevenir formación o
arrastre de contaminantes (por ejemplo, niveles de microorganismos objetables).
5.12 Para prevenir contaminación cruzada, los equipos no
dedicados deben limpiarse entre producción de diferentes materiales.
5.13 Deben definirse y justificarse los criterios de
aceptación para residuos, los procedimientos y agentes de limpieza.
5.14 El equipamiento debe identificarse por medios
apropiados respecto de su contenido y estadío de limpieza.
Calibración
5.15 El equipamiento de control, pesadas, medidas, monitoreo
y ensayos críticos para asegurar la calidad de los intermediarios o APIs, deben
ser calibrados acorde a procedimientos escritos y un programa establecido.
5.16 Las calibraciones deben realizarse utilizando
estándares trazables o certificados, si existiera.
5.17 Deben conservarse registro de estas calibraciones.
5.18 El estado de calibración actualizado de equipamiento
crítico debe ser conocido y verificado.
5.19 No deben utilizarse instrumentos que no posean
criterios de calibración.
5.20 Las desviaciones de estándares de calibración
aprobados en instrumentos críticos, debe investigarse para determinar el
impacto en la calidad de los intermediarios o APIs elaborados utilizando este
equipamiento después de la última calibración satisfactoria.
Sistemas Computarizados
5.21 Los sistemas computarizados relacionados con BPF
deben validarse. La profundidad y extensión de la validación depende de la
diversidad, complejidad y lo crítico de la aplicación computarizada.
5.22 La calificación de la instalación y la calificación
operacional debe demostrar la adecuación de hardware y software para realizar
la tarea asignada.
5.23 El software comercialmente disponible que ha sido
calificado no requiere el mismo nivel de testeo. Si el sistema que existe no
fue validado en la instalación, una validación retrospectiva puede llevarse a
cabo si está disponible la documentación adecuada.
5.24 Los sistemas computarizados deben tener suficientes
controles para prevenir acceso no autorizado o cambio de datos. Debe haber
control para prevenir omisiones en datos (por ejemplo, sistema desconectado y
datos no capturados). Debe haber registro de cualquier cambio de datos echo, el
ingreso previo, quien hizo el cambio y cuando.
5.25 Deben estar disponibles los procedimientos escritos
para la operación y mantenimiento de los sistemas computarizados.
5.26 Cuando se ingresan manualmente datos críticos debe
haber un control adicional en la exactitud de la entrada. Dicha operatoria
puede ser realizada por un segundo operador o por el mismo sistema.
5.27 Si se producen incidentes en sistemas computarizados
que puedan afectar la calidad de intermediarios o APIs, la confiabilidad de los
registros o resultados de análisis, deben registrarse e investigarse.
5.28 Los cambios en sistemas computarizados deben
realizarse de acuerdo a los cambios de procedimiento y deben ser formalmente
autorizados, documentados y testeados. Los registros deben llevarse para todos
los cambios, incluyendo modificaciones y mejoras hechas al hardware, software y
cualquier otro componente crítico del sistema. Estos registros deben demostrar
que el sistema se mantiene en estado de validación.
5.29 Si se produce una caída del sistema o el mismo falla,
lo cual conduce a una pérdida permanente de registros debe proveerse de un
sistema "back up". Para todo sistema computarizado debe establecerse
un medio de protección de aseguramiento de datos.
5.30 Los datos pueden ser registrados por un segundo medio
además del sistema computarizado.
6 DOCUMENTACION Y REGISTRO
Sistema de Documentación y Especificaciones
6.1 Todos los documentos relacionados con la elaboración
de intermediarios o APIs deben prepararse, revisarse, aprobarse y distribuirse
de acuerdo a procedimientos escritos. Tales documentos pueden estar en papel o
en forma electrónica.
6.2 La emisión, revisión, reemplazo y retiro de todos los
documentos debe ser controlada con mantenimiento de revisiones históricas.
6.3 Un procedimiento debe ser establecido para la
retención de todos lo documentos apropiados (por ejemplo, informes de
desarrollos históricos, de scale-up, de transferencia técnica y de procesos de
validación; registros de entrenamiento, de producción, de control y de
distribución). Los tiempos de retención de estos documentos deben
especificarse.
6.4 Todos los registros de control, de producción y
distribución deben ser retenidos por lo menos durante un año posterior a la
fecha de vencimiento del lote. En el caso de APIs con fecha de re-análisis los
registros deben mantenerse por lo menos durante tres años posteriores a la
distribución completa del lote.
6.5 Las entradas en los registros deben ser realizadas
indeleblemente en los espacios provistos para tal fin, inmediatamente después
de realizada la actividad e identificando a la persona que la realiza. Las
correcciones a dichas entradas deben poseer fecha y firma y permitir la lectura
del original.
6.6 Durante el período de retención los registros
originales o sus copias deben estar disponibles en el establecimiento donde se
desarrollan las actividades descriptas. Los registros pueden trasladarse a otro
sitio por medios electrónicos u otros.
6.7 Las especificaciones, instrucciones, procedimientos y
registros pueden retenerse como originales o copias certificadas tales como
fotocopias, microfilms, microfichas u otros. Cuando se utilizan técnicas de
reducción tales como microfilm o registro electrónico, debe encontrarse
disponible el equipamiento adecuado para obtener una copia segura.
6.8 Se deben establecer y documentar especificaciones para
material de partida, intermediarios cuando fuera necesario, APIs y materiales
de etiquetado y acondicionamiento. Además debe establecerse especificaciones
apropiadas para otros materiales como coadyuvantes de proceso, empacado u otros
materiales, utilizados durante la producción de intermediarios o APIs que
puedan impactar de forma crítica en la calidad de los mismos. Se deben
establecer y documentar criterios de aceptación para controles en proceso.
6.9 Si se usa firma electrónica en los documentos ésta
debe ser autenticada y segura.
Limpieza de Equipamiento y Registros de Uso
6.10 Los registros de uso, limpieza, sanitización y/o
esterilización y mantenimiento de los equipos principales deben llevar fecha y
hora (si corresponde), producto, y número de lote de cada lote procesado en el
equipo y la persona que ha realizado la limpieza y el mantenimiento del mismo.
6.11 Si el equipo está dedicado a la elaboración de un
intermediario o API, los registros individuales del equipo no son necesarios si
los lotes del intermediario o API siguen una secuencia trazable. En el caso en
que se utilicen equipos dedicados, los registros de limpieza, mantenimiento y
uso pueden formar parte del registro de lote.
Registros de Materiales de partida, Intermediarios,
Materiales de empaque y rotulado de APIs.
6.12 Los registros deben mantenerse incluyendo:
• El nombre del elaborador, identidad y cantidad de cada
despacho de cada lote de material de partida, intermediarios, o materiales de
empaque y rotulado para los APIs; el nombre del proveedor; el número de control
del proveedor, si se conoce, u otro número de identificación; el número
asignado y la fecha de recepción.
• Los resultados de cualquier test o análisis realizado y
las conclusiones derivadas de los mismos.
• Registros trazables del uso de materiales.
• Documentación correspondiente al examen y revisión de
los materiales de empaque y rotulado de APIs, en conformidad con las
especificaciones establecidas.
• La decisión final respecto al rechazo de materiales de
partida, materiales de empaque y rotulado de APIs y sus intermediarios.
6.13 Los rótulos maestros (aprobados) deben mantenerse
para ser comparados con los emitidos
Instrucciones Maestras de Producción (Registros Maestros
de Producción y Control)
6.14 Para asegurar la uniformidad de lote a lote, las
instrucciones maestras de producción para cada intermediario y API deben ser
preparadas, fechadas y firmadas por una persona, e independientemente
chequeadas, fechadas y firmadas por personal de la unidad de calidad.
6.15 Las instrucciones deben incluir:
• El nombre del intermediario o API a ser elaborado y un
código de identificación de referencia si corresponde.
• Una lista completa de materiales de partida, e
intermediarios designados por los nombres o códigos suficientemente específicos
para identificar cualquier característica especial de calidad.
• Un informe exacto de la cantidad o proporción de cada material
de partida o intermediario a ser usado, incluyendo la unidad de medida. Cuando
la cantidad no es fija, el cálculo para cada tamaño de lote debe incluirse. Se
deben justificar las variaciones en las cantidades.
• El área de elaboración y el equipamiento utilizado.
• Las instrucciones detalladas de producción deben
incluir:
- Secuencias a seguir.
- Especificaciones de proceso.
- Instrucciones de muestreo y controles en proceso con sus
criterios de aceptación cuando corresponda.
- Tiempos límites para completar etapas individuales del
proceso y/o el proceso total cuando corresponda.
- Rendimientos esperados en los tiempos y etapas del
proceso.
• Cuando sea necesario anotaciones especiales y
precauciones a seguir.
• Instrucciones para almacenamiento del intermediario o
API, para asegurar su aptitud de uso, incluyendo materiales de
acondicionamiento y rotulado, y condiciones y tiempos límites de
almacenamiento.
Registros de lote de Producción y Control
6.16 Los Registros de Producción deben prepararse para
cada intermediario y API y deben incluir información completa relacionada a la
producción y control de cada lote. El registro de cada lote de producción debe
controlarse antes de su emisión para asegurar que corresponda a la versión
correcta y sea una reproducción segura de las instrucciones maestras de
producción.
6.17 Los registros deben enumerarse con un único número de
identificación o lote, fechados y firmados al ser emitidos. En producción
continua, el código de producto junto con la fecha y la hora pueden servir como
única identificación hasta que el número final se asigne.
6.18 La documentación de cada etapa significativa del
proceso en el registro del lote de producción debe incluir:
• Fecha y tiempos.
• Identificación de los principales equipos (por ejemplo:
reactores secadores, mezcladoras, etc.) usados.
• Identificación específica de cada lote incluyendo pesos,
medidas, y número de lotes de materiales de partida, intermediarios o cualquier
material reprocesado usado durante la elaboración.
• Resultados reales registrados para parámetros críticos
de procesos.
• Muestreos realizados.
• Firma de las personas que llevan a cabo y supervisan o
chequean directamente cada etapa crítica en la operación.
• Resultados de análisis de laboratorio en proceso.
• Rendimiento real en etapas y tiempos del proceso.
• Descripción del empaque y rotulado para intermediarios o
APIs .
• Rótulos representativos del API o intermediario.
• Cualquier desviación detectada, su evaluación, su
investigación si corresponde o una referencia de que la misma se realiza
separadamente.
• Resultados del test de liberación.
6.19 Deben establecerse y seguirse procedimientos escritos
para las desviaciones críticas o fallas observadas en lotes de intermediarios o
APIs para cumplir especificaciones. La investigación debe extenderse a otros
lotes que puedan estar asociados con fallas o desviaciones específicas.
Registros de Laboratorio de Control
6.20 Los registros de laboratorio de control deben incluir
datos completos obtenidos de los análisis realizados para asegurar el
cumplimiento con los estándares y especificaciones establecidas, incluyendo:
• Una descripción de las muestras recibidas para ensayo,
incluyendo el nombre del material u origen, número de lote u otro código distintivo,
fecha de muestreo y, cuando corresponda, la cantidad y la fecha de recepción de
las mismas.
• Una referencia de cada método de análisis utilizado.
• Una referencia sobre pesadas y medidas de muestras
utilizadas para cada análisis según el método descripto; datos sobre
preparación y análisis de estándares de referencia, reactivos y soluciones
estándar.
• Un registro completo de los datos crudos generados en
cada análisis, cromatogramas, espectros y registros debidamente identificados
con el número de lote analizado.
• Hoja de cálculo incluyendo, por ejemplo unidades de
medida, factores de conversión y equivalencias.
• Informe de los resultados y su comparación con los
criterios de aceptación establecidos.
• Firma del analista y fecha en que se realizaron los
análisis.
• Fecha y firma de un supervisor.
6.21 Los informes completos deben mantenerse para:
• Cualquier modificación de un método analítico
establecido.
• Calibración periódica de instrumentos y aparatos.
• Ensayos de estabilidad realizados en APIs.
• Investigaciones sobre datos fuera de especificaciones.
Revisión de Registros de lote de producción.
6.22 Los procedimientos escritos deben establecerse y
seguirse para la revisión y aprobación de registros de lote de producción y
control de laboratorio, incluyendo empaque y rotulado, para determinar el
cumplimiento con las especificaciones establecidas del intermediario o API
antes de liberar el lote o distribuirlo.
6.23 Los registros de lote de producción y control de
laboratorio de etapas críticas del proceso deben ser revisados y aprobados por
la unidad de calidad antes que el lote de API sea liberado o distribuido. Los
correspondientes a etapas no críticas del proceso pueden ser revisados por
personal calificado de producción u otras unidades, siguiendo procedimientos
aprobados por la unidad de calidad.
6.24 Toda desviación, investigación e informe fuera de
especificaciones, debe ser revisado como parte del registro de lote previo a su
liberación.
6.25 La unidad de calidad puede delegar a la de
producción, la responsabilidad y autoridad para liberar intermediarios, excepto
en aquellos casos en que el intermediario se transfiera a un destino en el cual
se halla fuera del control del fabricante.
7- MANEJO DE MATERIALES
Controles Generales
7.1 Deben existir procedimientos que describan la
recepción, identificación, cuarentena, almacenamiento, manipulación, muestreo,
análisis y aprobación o rechazo de materiales.
7.2 Los elaboradores de intermediarios y/o APIs deben
poseer un sistema para la evaluación de suministros de materiales críticos.
7.3 Los materiales deben ser adquiridos de acuerdo a
especificaciones establecidas a proveedores aprobados por la unidad de calidad.
7.4 Si el proveedor de un material crítico no es el
elaborador, el productor del intermediario y/o API debe disponer del nombre y
la dirección del mismo.
7.5 Los cambios de proveedor de materiales de partida
críticos deben ser tratados de acuerdo a la sección 13, Control de Cambios.
Recepción y Cuarentena
7.6 Desde el ingreso y hasta su aprobación, cada
contenedor o grupo de contenedores de materiales debe ser examinado visualmente
para un correcto rotulado (incluida la correlación entre el nombre usado por el
proveedor y el utilizado en la empresa si fuesen diferentes), daños en el
contenedor, sellos rotos y evidencias de descomposición o contaminación. Los
materiales deben permanecer en cuarentena hasta haber sido muestreados,
analizados o examinados si corresponde y liberados para su uso.
7.7 Antes de que los materiales ingresados sean mezclados
con el stock existente (por ejemplo: solventes o stocks en silos) deben ser
identificados correctamente, analizados si corresponde, y liberados. Los
procedimientos deben estar disponibles para prevenir descarga errónea de
materiales en el stock existente.
7.8 Si los graneles recibidos se almacenan en tanques no
dedicados, debe asegurarse que no existe contaminación cruzada a partir de los
mismos. Para asegurar que esto no ocurra debe incluirse en forma total o
parcial la siguiente documentación:
- Certificado de limpieza
- Análisis de trazas de impurezas
- Auditorías del proveedor
7.9 Se deben identificar los contenedores principales y
sus correspondientes conductos, así como las líneas de llenado y descarga.
7.10 Cada contenedor de materiales o grupo de ellos, debe
identificarse con un código distintivo, lote o número de recepción. Dicho
número debe ser usado en el registro de disposición de cada lote. Debe existir
un sistema que permita identificar el estado de cada lote.
Muestreo y Análisis de Materiales de Producción Recibidos
7.11 Se debe realizar al menos un análisis para verificar
la identidad de cada lote de material, con excepción de los materiales
descriptos en 7.12. Un certificado de análisis del proveedor puede ser usado en
lugar de realizar otros ensayos, con tal que el elaborador posea un sistema
para la evaluación de sus proveedores.
7.12 La aprobación del proveedor debe incluir una
evaluación que aporte evidencia adecuada de que el elaborador puede proveer
consistentemente materiales que cumplan especificaciones. Se deben realizar
análisis completos sobre por lo menos tres lotes antes de reducir los análisis
internos. Sin embargo, como mínimo, un análisis completo debe ser llevado a
cabo a intervalos apropiados y comparado con el Certificado de Análisis. La
confiabilidad de los Certificados de Análisis debe chequearse a intervalos
regulares.
7.13 Los coadyuvantes de proceso, materiales de partida
altamente tóxicos o peligrosos, otros materiales especiales o materiales
transferidos a otra unidad dentro del control de la compañía, no requieren
análisis si el Certificado de Análisis del elaborador se obtiene, demostrando
que dichos materiales de partida cumplen con las especificaciones establecidas.
El examen visual de los contenedores, su rótulo y el registro de su número de
lote, debe ayudar en la identificación de dichos materiales. La ausencia de
análisis propios para estos materiales, debe justificarse y ducumentarse.
7.14 El muestreo del lote de material debe ser
representativo. El método de muestreo debe especificar el número de
contenedores a muestrear, que parte del contenedor es muestreada y la cantidad
de material a muestrear en cada contenedor. El número de contenedor a muestrear
y el tamaño de la muestra debe basarse en un plan de muestreo que tenga en
consideración lo critico del material, la variedad del material, los datos de
calidad históricos del proveedor y la cantidad necesaria para el análisis.
7.15 El muestreo debe realizarse en un ambiente definido y
con procedimientos diseñados para prevenir la contaminación del material
muestreado y de otros materiales.
7.16 Los contenedores a muestrear deben ser abiertos
cuidadosamente y posteriormente cerrados. Debe indicarse que el mismo ha sido
muestreado.
Almacenamiento
7.17 Los materiales deben manipularse y almacenarse de
manera de prevenir degradación, contaminación y contaminación cruzada.
7.18 Los materiales almacenados en tambores, cajas, o
bolsas, no deben situarse sobre el piso y, cuando corresponde, adecuadamente
separados para permitir su limpieza o inspección.
7.19 Los materiales deben almacenarse en condiciones y por
un período de tiempo que no afecte de manera adversa su calidad, y debe
controlarse que se utilice primero el stock más antiguo.
7.20 Ciertos materiales pueden almacenarse al aire libre
en contenedores adecuados, provistos de rótulos de identificación que
permanezcan legibles. Los contenedores deben limpiarse previamente a ser
abiertos para su uso.
7.21 Los materiales rechazados deben identificarse y
controlarse bajo un sistema de cuarentena diseñado para prevenir su uso en la
elaboración.
Re-evaluación
7.22 Cuando corresponda los materiales deben ser
re-evaluados para determinar la conveniencia de su uso (por ejemplo, luego de
un prolongado almacenamiento o exposición al calor o la humedad).
8. PRODUCCION Y CONTROLES EN PROCESO
Operaciones de Producción
8.1 Los materiales de partida para la elaboración de
intermediarios y APIs deben pesarse bajo condiciones que no afecten su
idoneidad. Los instrumentos de pesada y medida deben ser convenientemente
precisos para el uso propuesto.
8.2 Si el material es subdividido para su posterior uso en
operaciones de producción, los contenedores deben ser adecuados e identificarse
con la siguiente información:
• Nombre del material y/o código.
• Número de control o recepción.
• Peso o medida del material en el nuevo envase.
• Fecha de reanálisis o re-evaluación, si corresponde.
8.3 Las pesadas críticas, medidas u operaciones de subdivisión
del material deben estar sujetas a un control equivalente. Previo a su uso,
personal de producción debe verificar que los materiales son los especificados
en el registro de lote para el intermediario o API propuesto.
8.4 Otras actividades críticas deben estar sujetas a un
control equivalente.
8.5 Los rendimientos reales deben compararse con los
esperados en las distintas etapas de producción. Los rendimientos esperados y
sus respectivos rangos deben establecerse basados en pruebas de laboratorio,
escalas piloto o datos de manufactura. Las desviaciones en el rendimiento
asociadas con etapas críticas del proceso, deben investigarse para determinar
su impacto o potencial impacto en la calidad resultante de los lotes correspondientes.
8.6 Cualquier desviación debe documentarse, explicarse e
investigarse.
8.7 El estado de las unidades principales del equipamiento
de proceso debe indicarse, ya sea en las unidades individuales o por medio de
documentación apropiada, sistema computarizado u otros medios.
8.8 Los materiales a ser reprocesados deben controlarse
adecuadamente para prevenir su uso no autorizado.
Tiempos límites
8.9 Si se establece un tiempo límite en las instrucciones
maestras de producción (ver 6.14) el mismo debe asegurar la calidad de los
intermediarios y APIs. Las desviaciones deben ser documentadas y evaluadas. Los
tiempos límites pueden ser inapropiados cuando durante el proceso se deba
alcanzar un valor previsto para determinado parámetro (por ejemplo: ajuste de
pH, hidrogenación, secado hasta una especificación predeterminada). En este
caso, las etapas del proceso o la finalización de las reacciones deben
determinarse por muestreos y análisis en proceso.
8.10 Los intermediarios necesarios para procesos posteriores
deben almacenarse bajo condiciones apropiadas que aseguren su aptitud de uso.
Muestreo y Controles en Proceso
8.11 Se deben establecer procedimientos escritos que
permitan monitorear la evolución y el control de etapas del proceso, las cuales
puedan causar variabilidad en la calidad de intermediarios y APIs. Los
controles en proceso y sus criterios de aceptación deben definirse basándose en
la información obtenida durante las etapas de desarrollo o por datos
históricos.
8.12 El criterio de aceptación y el tipo y alcance de los
controles, dependerán de la naturaleza del intermediario o API que se esté
elaborando, de la reacción o etapa del proceso que se esté realizando y del
grado en que el proceso pueda producir una variación en la calidad del producto.
En las primeras etapas de proceso puede ser apropiado realizar controles en
proceso menos exigentes, mientras que controles más estrictos deben realizarse
en las últimas etapas (por ejemplo etapas de aislación, purificación).
8.13 La unidad(es) de calidad debe escribir y aprobar
controles críticos en proceso (y monitoreo de procesos críticos), incluyendo
puntos de control y métodos utilizados.
8.14 Los controles en proceso pueden ser realizados por
personal calificado del Departamento de producción. El proceso puede ser
ajustado sin aprobación previa por la unidad(es) de calidad, si el ajuste se
realiza dentro de los límites aprobados por la unidad(es) de calidad. Todos los
controles y resultados deben ser exhaustivamente documentados como parte del
registro de lote.
8.15 Deben existir procedimientos escritos para métodos de
muestreo de materiales en proceso, intermediarios y APIs. Los planes de
muestreo y procedimientos deben basarse en prácticas de muestreo
científicamente ciertas.
8.16 Los muestreos en proceso deben realizarse utilizando
procedimientos que eviten la contaminación del material muestreado. Los
procedimientos deben establecerse de manera de asegurar la integridad de la
muestra después de obtenida.
8.17 Normalmente no son necesarias investigaciones de
datos fuera de especificación para aquellos análisis en proceso realizados con
el propósito de monitorear y/o ajustar el mismo.
Mezcla de Lotes de Intermediarios o APIs
8.18 Para el propósito de este documento
"mezcla" se define como el proceso de combinar materiales dentro de
la misma especificación para producir un intermediario o API homogéneo. La
mezcla de fracciones en proceso de un lote en particular (por ejemplo
recolección de varias cargas de centrífuga a partir de un único lote de
cristalización) para su posterior procesamiento, se considera parte del proceso
de producción y no una mezcla.
8.19 Los lotes fuera de especificación no deben ser
mezclados con otros lotes con el propósito de cumplir especificaciones. Cada
lote incorporado en la mezcla debe ser elaborado usando el proceso establecido
y debe ser analizado individualmente corroborando que cumpla especificaciones
antes de ser incorporado a la mezcla.
8.20 Las operaciones aceptables en el mezclado incluyen,
pero no se limitan a:
• Mezclado de pequeños lotes para incrementar el tamaño de
lote;
• Mezcla de remanentes (por ejemplo pequeñas cantidades de
material aislado) provenientes de lotes del mismo intermediario o API para
formar un único lote.
8.21 Los procesos de mezclado deben ser adecuadamente
controlados y documentados, y el lote de la mezcla debe ser analizado debiendo
cumplir las especificaciones establecidas.
8.22 El registro de lotes del proceso de mezclado debe
permitir la trazabilidad retrospectiva hacia los lotes individuales que dieron
origen a la mezcla
8.23 Cuando los parámetros físicos de los APIs son
críticos (por ejemplo: APIs usados en formas sólidas orales o suspensiones),
las operaciones de mezclado deben validarse para demostrar homogeneidad del
lote obtenido. La validación debe incluir el análisis de parámetros críticos
(por ejemplo, distribución de tamaño de partícula, densidad del granel) que
pueden ser afectados.
8.24 Si el proceso de mezclado afecta la estabilidad,
deben realizarse controles adecuados al producto obtenido.
8.25 La fecha de expiración o de re-análisis del lote
obtenido por mezcla debe basarse en la fecha de elaboración del remanente o del
lote más antiguo utilizado en la mezcla.
Control de Contaminación
8.26 Si existe un control adecuado, los materiales
remanenetes pueden ser trabsferidos a sucesivos lotes del mismo intermediario o
API. Los ejemplos incluyen residuos adheridos a las paredes de un micronizador,
capa residual de cristales húmedos posterior a la descarga de centrífuga y una
descarga incompleta de fluidos o cristales provenientes de reactores hacia el
siguiente paso del proceso. Esta práctica no debe dar lugar a una transferencia
de degradados o de contaminación microbiana que pueda alterar el perfil de
impurezas establecido para el API.
8.27 Las operaciones de producción deben realizarse de
manera de prevenir la contaminación de intermediarios o APIs por otros
materiales.
8.28 Se deben tomar precauciones para evitar la
contaminación cuando se manipulan APIs posteriormente a su purificación.
9 EMPAQUE Y ROTULADO IDENTIFICATORIO DE APIS E
INTERMEDIARIOS
Generalidades
9.1 Deberán escribirse procedimientos que describan la
recepción, identificación, cuarentena, muestreo, evaluación, liberación y
manipuleo de los materiales de empaque y rotulado.
9.2 Dichos materiales deberán cumplir con especificaciones
establecidas. Los materiales que no las cumplan deberán ser rechazados a fin de
evitar su utilización accidental.
9.3 Deberán llevarse registros del movimiento de dichos
materiales sean éstos aceptados o rechazados, especificando su recepción y
evaluación.
Materiales de Empaque
9.4 El envase debe proveer protección adecuada del APIs o
Intermediarios contra deterioro o contaminaciones que pudieran darse durante su
transporte o almacenamiento.
9.5 El envase debe ser limpio y, cuando el producto lo
requiera, sanitizado en forma adecuada para el uso pretendido. No debe ser
reactivo, aditivo o absortivo como para alterarla calidad del producto dentro
de los límites especificados.
9.6 Si el envase es reutilizable, deberá ser limpiado
según procedimientos escritos.
Emisión y Control de Rótulos
9.7 El acceso a las áreas de rotulado estará restringido a
personal autorizado.
9.8 Deben usarse procedimientos para conciliar las
cantidades de materiales emitidos, utilizados y devueltos. Toda discrepancia
deberá ser investigada, y dicha investigación deberá ser aprobada por el
Departamento de Calidad.
9.9 Todo exceso de materiales ya impresos con un número de
lote, deberá ser destruido. Los materiales destinados a devolución deberán
almacenarse apropiadamente de forma de evitar confusiones.
9.10 Los rótulos obsoletos deberán ser destruidos.
9.11 El instrumental utilizado para la impresión de
rótulos debe ser controlado de forma de asegurar que toda impresión realizada
cumpla con las especificaciones del Registro de Producción de cada lote.
9.12 Los rótulos emitidos para un lote deben ser
examinados cuidadosamente a fin de verificar la conformidad con las especificaciones
del Registro Maestro de Producción. El resultado de dicha examinación deberá
ser documentado.
9.13 El Registro de Producción de cada lote deberá guardar
un ejemplar del rótulo correspondiente.
Operaciones de Empaque y Rotulado
9.14 Deberán existir procedimientos documentados a fin de
asegurar el uso de los materiales de empaque y rotulado apropiados.
9.15 Deberá prevenirse toda confusión durante las
operaciones de rotulado, por medio de una adecuada separación espacial entra
las operaciones relacionadas con distintos APIs o Intermediarios.
9.16 Los rótulos usados en los envases de APIs e
Intermediarios deben indicar el nombre o código identificatorio del producto,
el número de lote, y las condiciones de almacenamiento dado que dicha información
es crítica para asegurar la calidad del producto.
9.17 Si el APIs o Intermediarios se transferirá a algún
destino en el cual no se hallará bajo el control del fabricante, deberá
incluirse también en el rótulo los datos correspondientes a: nombre y dirección
del fabricante, cantidad de producto contenido, condiciones especiales de
transporte, y requerimientos legales especiales. Si el API posee una fecha de
vencimiento, ésta debe indicarse en el rótulo y en el Certificado de Análisis.
Si el producto posee una fecha de re-evaluación, ésta debe indicarse en el
rótulo y/o en el Certificado de Análisis.
9.18 Las instalaciones para el empaque y el rotulado deben
ser inspeccionadas inmediatamente antes de ser usadas, a fin de asegurar que
ningún material necesita ser cambiado para la próxima operación. El resultado
de dicha inspección deberá asentarse en los registros de producción o control
del lote.
9.19 Los APIs e Intermediarios empacados y rotulados deben
inspeccionarse a fin de verificar que los envases llevan el rótulo correcto. El
resultado de dicha inspección deberá asentarse en los registros de producción o
control del lote.
9.20 Los envases de APIs o Intermediarios que se
transferirán a algún destino en el cual no se hallarán bajo el control del
fabricante, deberán cerrarse herméticamente, de forma que si dicho cierre es
violado, el receptor estará alertado acerca de la posibilidad de que el
producto se halle alterado.
10- ALMACENAMIENTO Y DISTRIBUCION
Procedimientos de Almacenamiento
10.1 Las instalaciones deben ser adecuadas para el
depósito de todos los materiales en condiciones apropiadas (por ejemplo,
temperatura y humedad controladas cuando esto sea necesario). Deben llevarse
registros de dichas condiciones cuando estas sean críticas para el
mantenimiento de las características del producto.
10.2 A menos que exista un sistema alternativo para prevenir el
uso no autorizado o accidental de los productos correspondientes a cuarentena,
rechazo, devolución o recuperación, deberán asignarse áreas separadas de
almacenamiento temporario para dichos productos hasta que se tome la decisión
sobre el destino que se les dará.
Procedimientos de Distribución
10.3 Los APIs e Intermediarios sólo podrán ser liberados
para su distribución a terceros cuando el Departamento de Calidad los haya
liberado. Los APIs e Intermediarios en cuarentena podrán ser transferidos a
hacia otros Departamentos bajo el control de la empresa cuando el Departamento
de Calidad así lo autorice, y si los controles y documentación están en regla.
10.4 Los APIs e Intermediarios deben transportarse de
forma de no afectar su calidad.
10.5 Las condiciones especiales de transporte o
almacenamiento deben constar en el rótulo de los APIs e Intermediarios que así
lo requieran.
10.6 El elaborador debe asegurar que el contratista de
transporte para los APIs e Intermediarios conozca y cumpla las condiciones de
almacenamiento y transporte adecuadas.
10.7 Debe existir un sistema de registros de distribución
que permita la recuperación de cada lote de APIs y/o Intermediarios.
11 CONTROLES DE LABORATORIO
Controles Generales
11.1 Los Departamentos de Calidad independientes deben
tener a su disposición instalaciones de laboratorio adecuadas.
11.2 Deberán existir procedimientos documentados para el
muestreo, la evaluación, la aprobación o rechazo, y el registro y archivo de
los datos de laboratorio. Los registros del laboratorio deben llevarse de
acuerdo con la Sección "Registros de Laboratorio de Control".
11.3 Las especificaciones, técnicas de muestreo y
procedimientos de evaluación deben ser apropiados y con fundamento científico,
a fin de asegurar que las materias primas, loa APIs, los Intermediarios y los
materiales de rótulo y empaque cumplen con los estándares de calidad y/o
pureza. Las especificaciones y procedimientos de evaluación deben ser
consistentes con aquellos registrados en los archivos. Podrán existir
especificaciones adicionales a las incluidas en los archivos. Las
especificaciones, técnicas de muestreo y procedimientos de evaluación,
incluidos los cambios en ellos, deben ser esbozados por una unidad
organizacional adecuada, y aprobados por el Departamento de Calidad.
11.4 Las especificaciones para los APIs deben ser
establecidas de acuerdo con estándares aceptados, y consistentes con los
procesos de manufactura. Ellas deben incluir un control de impurezas (por
ejemplo, impurezas orgánicas e inorgánicas, y residuos de solventes). Si los
APIs tienen una especificación de pureza microbiológica, deberán establecerse y
cumplirse límites apropiados para el recuento de microorganismos, y ausencia de
microorganismos objetables. Si los APIs tienen una especificación para
endotoxinas, deberán establecerse y cumplirse límites apropiados.
11.5 Los controles de laboratorio deben documentarse en el
momento de su ejecución. Toda desviación de los procedimientos descriptos
deberá documentarse y explicarse.
11.6 Todo resultado fuera de especificación (OOS) deberá
ser investigado y documentado de acuerdo con un procedimiento. Dicho
procedimiento requerirá análisis de datos, evaluación de la existencia de algún
problema de relevancia, designación de acciones correctivas, y conclusiones.
Todo remuestreo y/o re-evaluación tras un OOS debe realizarse según un
procedimiento documentado.
11.7 Los reactivos y soluciones estándar deben prepararse
y rotularse siguiendo procedimientos escritos. Deberá establecerse
adecuadamente una fecha de expiración para reactivos y soluciones estándar.
11.8 Para la elaboración de APIs se deberá contar con un
Estándar de Referencia Primario adecuado, cuya fuente deberá estar documentada.
Deberán llevarse registros del almacenamiento y el uso de cada Estándar de
Referencia Primario, de acuerdo con las recomendaciones del proveedor. Los
Estándares de Referencia Primario provistos por una fuente reconocida
oficialmente se usan normalmente sin evaluación previa, siempre que se hayan
mantenido bajo las condiciones recomendadas por el proveedor.
11.9 Cuando no se dispone un Estándar de Referencia
Primario de una fuente reconocida oficialmente, deberá establecerse un
"Estándar Primario de la casa". Este deberá evaluarse adecuadamente
para establecer totalmente su identidad y pureza. Dicha evaluación se
documentará apropiadamente.
11.10 Se deberá preparar, identificar, evaluar, aprobar y
almacenar un Estándar de Referencia Secundario. La aptitud de cada lote del
Estándar de Referencia Secundario debe determinarse antes de ser usado por
primera vez, comparándolo con el Estándar de Referencia Primario. Además, cada
lote deberá ser re-evaluado periódicamente según un protocolo escrito.
Evaluación de APIs e Intermediarios
11.11 Sobre cada lote de APIs e intermediarios se
realizarán ensayos de laboratorio adecuados para determinar su cumplimiento con
las especificaciones.
11.12 Para cada APIs se establecerá un perfil de impurezas
describiendo las impurezas identificadas o no en un típico lote, producido bajo
un proceso de producción controlado específico. Dicho perfil de impurezas algún
dato analítico de identidad (por ejemplo, tiempo de retención), el rango de
cada impureza observada, y la clasificación de cada impureza identificada (por
ejemplo, orgánica, inorgánica, solvente). Normalmente, el perfil de impurezas
es dependiente del proceso de producción y del origen del APIs. El perfil de impurezas
no suele ser necesario para APIs de origen vegetal o provenientes de tejidos
animales. Las consideraciones biotecnológicas se hallan descriptas en ICH
Guideline Q6B.
11.13 El perfil de impurezas debe ser comparado, a
intervalos adecuados, contra los datos históricos, a fin de detectar cambios en
el APIs provenientes de modificaciones en la material de partida, en parámetros
operativos de los equipos, o en el proceso de producción.
11.14 Cuando exista una especificación de calidad
microbiológica, sobre cada lote de APIs e Intermediarios deberán realizarse los
ensayos microbiológicos apropiados.
Validación de Procedimientos Analíticos (ver sección 12)
Certificados de Análisis
11.15 Para cada lote de APIs o intermediarios deberá
emitirse un Certificados de Análisis auténtico cuando este sea solicitado.
11.16 El Certificados de Análisis deberá proveer
información acerca de nombre del APIs o intermediarios, número de lote, grado y
fecha de liberación cuando éstos correspondan. Para APIs o intermediarios que
posean fecha de vencimiento, ésta debe constar en el Certificado de Análisis y
en el rótulo. Para APIs o Intermediarios que posean fecha de re-evaluación,
ésta debe constar en el Certificado de Análisis y/o en el rótulo.
11.17 El Certificado de Análisis debe incluir cada
evaluación desarrollada de acuerdo los requerimientos del cliente, incluyendo
los límites de aceptación, y los resultados numéricos obtenidos cuando
correspondan.
11.18 El Certificado de Análisis deberá estar fechado y
firmado por personal autorizado del Departamento de Calidad, y en él debe
constar el nombre, dirección y teléfono del elaborador original. Cuando el
análisis lo haya llevado a cabo un reacondicionador o reprocesador, los datos
(nombre, dirección y teléfono) que deberán constar el Certificado de Análisis
serán los de este último, y se añadirá además una referencia con el nombre del
elaborador original.
11.19 Si un nuevo Certificado de Análisis fuera emitido
por o en nombre de un reacondicionador, reprocesador o agente, el Certificado
deberá llevar los datos (nombre, dirección y teléfono) del laboratorio que
realizó los análisis. Además deberá incluir una referencia con el nombre del
elaborador original, y una copia del Certificado de Análisis original.
Monitoreo de la Estabilidad de APIs
11.20 Debe diseñarse un Programa documentado de Monitoreo
"on-going" a fin de controlar las características de estabilidad de
los APIs. Los resultados se utilizarán para confirmar las condiciones
apropiadas de almacenamiento, y las fechas de re-evaluación y expiración.
11.21 Los procedimientos usados en la evaluación de
estabilidad deben estar validados y ser indicadores de la estabilidad.
11.22 Las muestras para evaluación de estabilidad deben
almacenarse en envases que simulen o se asemejen a los envases de venta.
11.23 Normalmente, los primeros tres lotes comerciales de
APIs elaborados ingresan al Programa de Monitoreo para confirmar las fechas de
re-evaluación y expiración. De cualquier forma, cuando los datos de estudios
previos indiquen que el APIs es probablemente estable por al menos dos años,
podrán ingresarse al Programa menos de tres lotes.
11.24 A partir de entonces, se agregará al Programa al menos un
lote de APIs por año (salvo que no se haya elaborado ninguno) para confirmar
los datos de estabilidad.
11.25 En el caso de productos con vida estable corta, la
evaluación debe hacerse más frecuentemente. Por ejemplo, en el caso de
productos biotecnológicos o biológicos cuya vida estable suele ser de un año o
menos, la evaluación debe hacerse mensualmente durante los primeros tres meses,
y a partir de ahí, a intervalos de tres meses. Cuando existan datos que
confirmen que la estabilidad del producto no está comprometida, se podrá
considerar la eliminación o modificación de algunos intervalos.
11.26 Cuando corresponda, las condiciones estables de
almacenamiento deberán ser consistentes con la ICH Guideline de estabilidad.
Fechas de Re-evaluación y Expiración
11.27 Cuando un producto se transferirá fuera del control
del sistema de control de materiales del elaborador, y se le asigna una fecha
de re-evaluación, deberá disponer de la información correspondiente a
estabilidad (por ejemplo, fecha de publicación, resultados de la evaluación).
11.28 La fecha de re-evaluación o expiración deberá estar
basada en la evaluación de datos obtenidos de estudios de estabilidad. Es de
uso más común la fecha de re-evaluación que la de expiración.
11.29 Pueden obtenerse datos preliminares de la fecha de
re-evaluación o expiración a partir de lotes a escala piloto si: (1) El método
y procedimiento de manufactura del lote piloto simulan el proceso final a
utilizarse en la manufactura a escala comercial; y (2)La calidad del producto
obtenido es representativa de aquel que se obtendrá a escala comercial.
11.30 Deberá guardarse una muestra representativa que
permita desarrollar le re-evaluación cuando corresponda.
Muestras de Retención o Reserva
11.31 La finalidad de la toma de Muestras de Retención es
permitir potenciales evaluaciones futuras de la calidad de los lotes de APIs o
intermediarios. No se utilizarán para futuras evaluaciones de estabilidad.
11.32 Las Muestras de Retención se guardarán,
adecuadamente identificadas, hasta un año posterior a la fecha de expiración
establecida por el elaborador, o hasta tres años posteriores a la fecha de
distribución (entre ambos, se tomará el plazo más largo). Para APIs con fecha
de re-evaluación, se tomarán Muestras de Retención similares, las cuales se
guardarán por tres años tras la completa distribución por parte del elaborador.
11.33 Las Muestras de Retención deben almacenarse con el
mismo sistema de empaque con el que se almacenen los APIs a comercializar, o de
alguna forma que lo proteja aún mejor. La cantidad de producto tomado como Muestras
de Retención deberá ser suficiente como para permitir realizar al menos dos
análisis completos según se describan en la monografía de la Farmacopea, o, de no hallarse descriptos, dos análisis completos según las especificaciones.
12 VALIDACION
Política en Validación
12.1 La política general, intenciones y puntos de vista de
la compañía en relación con Validación (incluyendo validación de procesos de
producción, procedimientos de limpieza, métodos analíticos, controles durante
los procesos, sistemas computarizados y personal responsable del diseño,
revisión, aprobación y documentación de cada etapa de Validación), deberá estar
documentada.
12.2 Los parámetros críticos deberán estar identificados a
partir de la fase de desarrollo o de datos históricos, y deberán estar
definidos los lineamientos necesarios para que las operaciones sean
reproducibles. Esto incluirá:
- Definir el APIs en término de sus atributos críticos.
- Identificar los parámetros del proceso que pudieran
afectar la calidad de dichos atributos críticos.
- Determinar los lineamientos para cada parámetro crítico
del proceso que se utilizará rutinariamente durante la manufactura y el control
del proceso.
12.3 La Validación deberá extenderse a aquellas
operaciones que son críticas para la calidad y la pureza de los APIs.
Documentación de la Validación
12.4 Deberá establecerse un protocolo de Validación
escrito que especifique cómo debe llevarse a cabo la Validación de cada proceso en particular. Dicho protocolo deberá estar revisado y aprobado
por el Departamento de Calidad y por algún otro Departamento designado.
12.5 EL protocolo de Validación deberá especificar los
pasos críticos del proceso y los criterios de aceptación, así como el tipo de
Validación que se llevará a cabo (por ejemplo, Retrospectiva, Prospectiva,
Concurrente), y el número de corridas del proceso.
12.6 Deberá realizarse un reporte de Validación que resuma
los resultados obtenidos, que comente toda desviación observada y exprese las
conclusiones apropiadas, incluyendo las modificaciones recomendadas para
corregir las deficiencias existentes.
12.7 Toda variación del protocolo de Validación deberá
documentarse con la justificación correspondiente.
Calificación
12.8 Antes de comenzar con las actividades relacionadas
con los procesos de Validación, se deberá llevar a cabo la Calificación de los equipos críticos y de los sistemas auxiliares. La Calificación normalmente se lleva a cabo realizando las siguientes actividades, en forma
individual o combinada:
- DQ (Calificación del Diseño): verificación documentada
de que el diseño propuesto de instalaciones, equipamientos o sistemas es
adecuado para su propósito.
- IQ (Calificación de las Instalaciones): verificación
documentada de que los equipamientos o sistemas, tal como se hallan instalados
o con alguna modificación, cumplen tanto con el diseño aprobado, como con las
recomendaciones del fabricante y/o con los requerimientos del usuario.
- OQ (Calificación Operacional): verificación documentada
de que los equipamientos o sistemas, tal como se hallan instalados o con alguna
modificación, se desempeñan como es deseado en todos los ámbitos en los que se
anticipa que operarán.
- PQ (Calificación de Desempeño): verificación documentada
de que los equipamientos y sistemas auxiliares, tal como se hallan instalados y
conectados entre sí, pueden desempeñarse en forma eficiente y reproducible,
basándose en los procesos y especificaciones aprobadas.
Enfoques del Proceso de Validación
12.9 El Proceso de Validación es la evidencia documentada
de que el proceso, operado según los parámetros establecidos, puede
desempeñarse en forma eficiente y reproducible para producir el APIs o
intermediarios cumpliendo con las especificaciones y los atributos de calidad
predeterminados.
12.10 Existen tres enfoques para la Validación. El enfoque preferido es el Prospectivo, pero hay casos en los que se pueden usar
otros enfoques, detallándose estos a continuación.
12.11 La Validación Prospectiva se lleva a cabo normalmente en todos los procesos para APIs según 12.2. La Validación Prospectiva sobre un proceso para APIs debe estar finalizada antes de la
distribución comercial del producto terminado manufacturado a partir de dicho
APIs.
12.12 La Validación Concurrente puede realizase cuando no se dispone de suficientes datos replicados de la producción de distintos lotes de
APIs, a causa de la elaboración de un número limitado de lotes, o de la
producción poco frecuente, o de la producción a través de un proceso validado
diferente. La distribución comercial del producto terminado manufacturado a
partir de este APIs podrá realizarse antes de que se concluya la Validación Concurrente, basándose en la minuciosa evaluación y monitoreo de los lotes del
APIs.
12.13 La Validación Retrospectiva podrá usarse en casos de excepción, para procesos en los que está
bien establecido que no se han detectado cambios significativos en la calidad
del APIs cuando en su elaboración hubo alguna variación en la material de
partida, los equipamientos, los sistemas, las instalaciones, o el proceso de
producción. Este enfoque podrá usarse si se cumple con todos los siguientes
ítems:
- Se hallan identificados los atributos críticos de
calidad y los parámetros críticos del proceso.
- Se han establecido controles y criterios de aceptación
apropiados durante el proceso.
- No ha habido fallas significantes en el producto o en el
proceso por causas distintas a errores del operador o fallas del equipo sin
relación con la adecuabilidad de éste.
- Se han establecido perfiles de impurezas para el APIs.
12.14 Los lotes sobre los que se realizará una Validación
Retrospectiva deberán ser representativos de todos los lotes fabricados durante
el período en estudio (incluyendo los lotes que han quedado fuera de
especificación), y el número de lotes utilizado deberá ser suficiente como para
demostrar la consistencia del proceso. Las muestras de retención pueden ser
utilizadas para evaluar el proceso en forma retrospectiva.
Programa de Validación de Procesos
12.15 El número de corridas del proceso necesarias para la Validación dependerá de la complejidad del proceso o de la magnitud del cambio en el proceso.
Para la Validación Prospectiva y la Concurrente, pueden usarse tres lotes sucesivos producidos exitosamente, pero puede haber casos en los que puede
necesitarse un número mayor a causa de la complejidad del proceso. Para la Validación Retrospectiva generalmente se analizan los datos de diez a treinta lotes
consecutivos, pero podrán analizarse menos si se justifica apropiadamente.
12.16 Los parámetros críticos del proceso deben
controlarse y monitorearse durante los estudios de Validación. Los parámetros
no relacionados con la calidad no precisan ser incluidos en la Validación.
12.17 La Validación debe confirmar que el perfil de
impurezas papa cada APIs se encuentra dentro de los límites especificados. El
perfil de impurezas debe ser similar o mejor que los datos históricos, cuando
corresponda, se lo usará para estudios clínicos y toxicológicos.
Revisión Periódica de los Sistemas Validados
12.18 Los sistemas y los procesos deberán ser evaluados
periódicamente para verificar que continúan trabajando en forma validada.
Cuando no se verifiquen cambios significativos y se confirme que se está
trabajando en forma consistente y dentro de las especificaciones, no será
necesario realizar una revalidación.
Validación de la Limpieza
12.19 Los procedimientos de limpieza normalmente deben ser
validados. Esta Validación debe a las situaciones o etapas del proceso donde la
contaminación de los materiales representa un gran riesgo para la calidad del
APIs. (por ejemplo, al inicio de la producción es importante evaluar la
remoción de residuos anteriores).
12.20 La Validación de la limpieza debe reflejar el patrón
de uso de los equipos. Si distintos APIs o Intermediarios se elaboran en el
mismo equipo y la limpieza de éste se realiza de igual forma, se elegirá un
producto representativo para la validación de la limpieza. Dicha elección se
basará en la solubilidad, la dificultad de la limpieza, y en el cálculo del
límite de residuos basado en la potencia, la toxicidad, y la estabilidad.
12.21 El protocolo de Validación de limpieza debe
describir el equipo a ser limpiado, el procedimiento, los materiales, los
niveles aceptables de limpieza, los parámetros a monitorear y los métodos
analíticos. También debe indicar el tipo de muestras a tomar y como deben éstas
ser tomadas y rotuladas.
12.22 El muestreo debe incluir frotado, enjuagado u otro
método adecuado para detectar residuos solubles e insolubles. La técnica debe
permitir evaluar cuantitativamente los niveles de los residuos remanentes en
las superficies del equipo después de ser limpiado. El muestreo por frotado
será impracticable en los casos en que la superficie en contacto con el
producto es inaccesible.
12.23 Deberán usarse métodos analíticos validados con
sensibilidad como para detectar residuos o contaminantes. Deberá establecerse
el nivel de recuperación obtenido con ese método. El límite de residuos deberá
ser alcanzable, verificable, práctico, y basado en el residuo más retirable. El
límite se podrá establecer basándose en la mínima cantidad de APIs con
actividad farmacológica, toxicológica, o fisiológica conocida.
12.24 En aquellos procesos en los que el APIs posee
límites microbiológicos o de endotoxinas, los estudios de limpieza o
sanitización de los equipos deben apuntar a dichas contaminaciones.
12.25 Tras la Validación, los procedimientos de limpieza deben ser monitoreados cada intervalos adecuados a fin de asegurar que se
están realizando eficientemente. La limpieza de los equipos puede verificarse
por ensayos analíticos y por examinación visual, cuando sea posible.
Validación de Métodos Analíticos
12.26 Los métodos analíticos deberán ser validados a menos
que la técnica empleada esté incluida en la Farmacopea o en algún otro Estándar de Referencia reconocido. No obstante, la aptitud de
todos los métodos de evaluación usados debe ser verificada y documentada bajo
las condiciones de uso actuales.
12.27 Los métodos analíticos deberán ser validados
considerando las características incluidas en la ICH Guideline acerca de validación de métodos analíticos. El grado de validación desarrollado
deberá reflejar el propósito del análisis y la etapa del proceso de producción
del APIs.
12.28 Debe considerarse que los equipos estén
adecuadamente calificados antes de comenzar con la Validación de los métodos analíticos.
12.29 Deberán llevarse registros completos de toda
modificación hecha sobre un método analítico validado. Dichos registros deberán
incluirla causa de la modificación y los datos adecuados para verificar que la
modificación genera resultados que son certeros y confiables.
13 CONTROL DE CAMBIOS
13.1 Deberá establecerse un sistema formal de control de
cambios para evaluar todo cambio que pueda afectar la producción y el control
de un API o intermediario.
13.2 Deberán existir procedimientos escritos para la
identificación, documentación, revisión y aprobación de cambios en las materias
primas, en las especificaciones, en los métodos analíticos, en las
instalaciones, en los sistemas soportes, en los equipos (incluyendo el
hardware), en las etapas del procesos, en el empaque y rotulado, y en el
software.
13.3 Toda propuesta acerca de cambios relevantes en las
GMP debe ser esbozada, revisado y aprobado por la unidad organizacional
adecuada, y además será revisado y aprobado por el Departamento de Calidad.
13.4 Se deberá evaluar el impacto potencial del cambio
propuesto sobre la calidad del API o el intermediario. Para determinar el nivel
de evaluación, validación y documentación necesarios para justificar el cambio
en un proceso validado, puede ser de ayuda un procedimiento de clasificación.
Los cambios pueden clasificarse (por ejemplo, menor o mayor) dependiendo de la
naturaleza y la extensión de los cambios, y de los efectos que esos cambios
pueden causar en el proceso. Las evaluaciones y estudios validados adicionales
necesarios para justificar el cambio se determinarán según juicio científico.
13.5 Cuando se implementen los cambios aprobados se
tomarán medidas para asegurarse de que todos los documentos afectados por el
cambio fueron revisados.
13.6 Tras la implementación del cambio deberá realizarse
una evaluación sobre los primeros lotes producidos.
13.7 En el caso de cambios críticos deberá evaluarse el
impacto potencial sobre las fechas de reevaluación y expiración establecidas.
De ser necesario se tomarán muestras del APIs o I producido bajo el proceso ya
modificado las cuales podrán ingresar en un programa de estudio de estabilidad
acelerado y/o ser agregados al programa de monitoreo de estabilidad.
13.8 Los elaboradores de la forma de dosificación
corriente deben ser notificados de los cambios en los procesos de producción y
de control que puedan impactar en la calidad del APIs.
14 RECHAZO Y REUTILIZACION DE MATERIALES
Rechazo
14.1 Los APIs e intermediarios que no cumplieran con las
especificaciones deberán ser apropiadamente identificados y puestos en
cuarentena. Dichos productos podrán ser reprocesados o reelaborados según se
describe a continuación. El destino final de los productos rechazados deberá
ser documentado.
Reprocesamiento
14.2 Normalmente se considera como aceptable la
reinserción dentro del proceso de un API o intermediario (incluyendo los que no
cumplen con las especificaciones) y su reprocesamiento por medio de la
repetición de un paso de recristalización u otro paso físico o químico (por
ejemplo, destilación, filtración, cromatografía, molienda). Sin embargo, si
dicho reprocesamiento se usa en la mayoría de los lotes, el proceso deberá
incluirse dentro del proceso estándar de elaboración.
14.3 La continuación de una etapa que se hallaba en curso
tras un control en proceso que mostró que esta etapa se hallaba incompleta se
considerará como parte del proceso normal y no como un reprocesamiento.
14.4 La reinserción dentro del proceso de una materia que
no reaccionó y la repetición de la reacción química se considerará como un
reprocesamiento a menos que sea parte del proceso establecido. Dicho reprocesamiento
deberá estar precedido por una evaluación cuidadosa a fin de asegurar que la
calidad del API o intermediario no se verá modificada por la posible formación
de productos relacionados o sustancias que han reaccionado en exceso.
Re-elaboración
14.5 Antes de tomar la decisión de reelaborar un lote que
no cumple con las especificaciones deberá realizarse una investigación acerca
de la razón por la cual se dio dicho incumplimiento.
14.6 Los lotes que han sido re-elaborados deberán ser adecuadamente
evaluados y documentados a fin de verificar que su calidad es equivalente a la
de los lotes elaborados normalmente. Para los procesos de re-elaboración suele
ser adecuada una Validación Concurrente. Deberá escribirse un protocolo que
defina el procedimiento de re-elaboración, que lo describa y especifique los
resultados esperados. Si sólo es un lote el que debió ser re-elaborado, será
suficiente con escribir un reporte, y la liberación se autorizará una vez que
el lote se evalúe como aceptable.
14.7 Debe contarse con un perfil de impurezas que permita
comparar el lote re-elaborado con los lotes obtenidos por el proceso
establecido. Cuando los métodos analíticos no sean adecuados para caracterizar
le lote re-elaborado, deberán usarse métodos adicionales.
Recuperación de Materiales y Solventes
14.8 La recuperación de reactivos, de APIs o los
intermediarios (por ejemplo, de una solución madre o de un filtrado) se
considera aceptable siempre que existan procedimientos aprobados y que los
productos recuperados cumplan con las especificaciones adecuadas para lo que se
los va a utilizar.
14.9 Los solventes pueden recuperarse y ser usados en los
mismos procesos o en procesos diferentes, siempre que, antes de ser usados o
mezclados con otros solventes aprobados, se realicen controles sobre los
procedimientos de recuperación que aseguren que los solventes cumple con los
estándares adecuados.
14.10 Los reactivos o solventes frescos y recuperados
podrán ser mezclados si el control apropiado indica que cumple con los
requisitos para lo que se los utilizará.
14.11 El uso de materiales recuperados deberá estar
adecuadamente documentado.
Devoluciones
14.12 Los APIs o intermediarios que se devolverán deberán
ser apropiadamente identificados y puestos en cuarentena.
14.13 Si las condiciones en las que los productos a
devolver han sido almacenados o transportados, antes o durante su retorno,
generan dudas acerca de su calidad, dichos productos deberán ser reprocesados
reelaborados o destruidos según corresponda.
14.14 Deberán llevarse registros de los APIs o
Intermediarios devueltos. Por cada devolución, la documentación deberá incluir:
- Nombre y dirección del depositario
- Nombre del APIs o Intermediario, número de lote, y
cantidad devuelta.
- Razón de la devolución.
- Uso o destino del APIs o Intermediario devuelto.
Reclamos y Retiro del Mercado
14.15 Todo reclamo relacionado con la calidad, ya sea oral
o escrito, deberá ser registrado e investigado según un procedimiento escrito.
14.16 El registro de un reclamo debe incluir:
- Nombre y dirección del reclamante;
- Nombre, título y teléfono de la persona que presentó el
reclamo;
- Naturaleza del reclamo (incluyendo nombre y lote del
API);
- Fecha de recepción del reclamo;
- Acción tomada inicialmente (incluyendo fechas e
identidad de la persona que inició la acción);
- Toda acción subsiguiente;
- Respuesta dada al reclamante;
- Decisión final tomada sobre el lote del API.
14.17 Se llevarán registros de los reclamos que permitirán
analizar tendencias, frecuencias relacionadas con el producto, y severidad; a
fin de tomar medidas correctivas inmediatas de ser necesario.
14.18 Deberán existir procedimientos escritos que definan
bajo qué circunstancias se considerará realizar un Retiro del mercado.
14.19 El procedimiento del Retiro del mercado debe
designar quién será responsable de evaluar la información, cómo se iniciará el
Retiro del mercado, quién debe ser informado acerca del Retiro del mercado, y
cómo se tratará el material recuperado.
14.20 Ante una situación grave o de potencial riesgo de
vida, las autoridades locales, nacionales o internacionales deberán ser
informadas y se les solicitará consejo al respecto.
15 ELABORADORES CONTRATADOS (INCLUYENDO LABORATORIOS)
15.1 Todos los elaboradores contratados deben cumplir con
las normas BPF definidas en esta guía. Debe tenerse especial consideración
acerca de la prevención de contaminaciones cruzadas y el seguimiento de la
trazabilidad.
15.2 Los elaboradores contratados deben ser evaluados por
el contratante acerca del cumplimiento de las BPF en las operaciones
específicas que se llevan a cabo en le sitio contratado.
15.3 Debe existir un contrato escrito y aprobado o acuerdo
formal entre contratante y contratado que defina en detalle las responsabilidades
de cada parte acerca de las BPF.
15.4 El contrato debe permitir que el contratante audite
las instalaciones del contratado en relación con el cumplimiento de las BPF.
15.5 Cuando se autoriza la existencia de un subcontratado,
el contratado no debe confiarle a aquél más de la tercera parte del trabajo que
se le confió, sin la evaluación y aprobación del contratante.
15.6 Deberán llevarse registros del elaborador o el
laboratorio en el sitio en el que se desarrollan las actividades, y estos deberán
estar siempre disponibles para ser consultados.
15.7 No se podrá realizar ningún cambio en los procesos,
equipamientos, métodos de evaluación, especificaciones, u otros requerimientos
contractuales sin la aprobación previa del contratante.
16 AGENTES, CORREDORES, COMERCIANTES, DISTRIBUIDORES, Y
REACONDICIONADORES.
Aplicación
16.1 Esta sección se aplicará a toda parte distinta del
elaborador original, que pueda comercializar y/o poseer, reenvasar, rerotular,
manipular, distribuir o almacenar APIs o Intermediarios.
16.2 Todos los agentes, corredores, comerciantes,
distribuidores, y reacondicionadores deberán cumplir con las normas BPF según
se describe en esta guía.
16.3 Trazabilidad de los APIs e Intermediarios
Distribuidos
16.4 Los agentes, corredores, comerciantes,
distribuidores, y reacondicionadores deberán mantener una completa trazabilidad
de los productos que distribuyen. Esta documentación deberá hallarse siempre
disponible, y deberá incluir:
- Identidad y dirección del elaborador original;
- Orden de compra;
- Documento de transporte;
- Documento de recepción;
- Nombre del APIs o Intermediario;
- Número de lote del fabricante;
- Registros de transporte y distribución;
- Todos los certificados de análisis auténticos,
incluyendo los del elaborador original;
- Fecha de re-evaluación o expiración.
Gerencia de Calidad
16.5 Los agentes, corredores, comerciantes,
distribuidores, y reacondicionadores deberán establecer, documentar e
implementar un sistema efectivo de gerenciamiento de la calidad, según se
especifica en la sección 2.
Reempaque, Rerotulado y Posesión de APIs e Intermediarios
16.6 El reempaque, rerotulado y posesión de APIs e
Intermediarios debe llevarse acabo bajo las adecuadas normas BPF, según consta
en esta guía, a fin de evitar confusiones o pérdidas de la pureza o la
identidad de APIs e Intermediarios.
16.7 El reempaque debe realizarse bajo las adecuadas
condiciones ambientales a fin de evitar contaminaciones comunes o cruzadas.
Estabilidad
16.8 Si un API o Intermediario es reempacado en un tipo de
envase diferente del usado por el elaborador original, deberán realizarse
estudios que verifiquen las fecha de re-evaluación o expiración asignada.
Transferencia de Información
16.9 Los agentes, corredores, comerciantes,
distribuidores, y reacondicionadores deberán transferir al cliente toda la
información regulatoria o relacionada con la calidad del producto recibida del
elaborador o un intermediario, así como del cliente al elaborador o intermediario.
16.10 Los agentes, corredores, comerciantes,
distribuidores, y reacondicionadores que provean APIs o Intermediarios a un
cliente, deberán informarle el nombre y el número de lote del producto
provisto.
16.11 Los agentes, corredores, comerciantes, distribuidores,
y reacondicionadores también deberán informar la identidad del elaborador
original a las autoridades regulatorias cuando así lo requieran. El elaborador
original puede responder ante las autoridades regulatorias en forma directa o a
través de agentes autorizados por el elaborador, dependiendo de la relación
entre el elaborador y dichos agentes.
16.12 Deberá cumplirse con la guía específica para
Certificados de Análisis incluida en la sección "Certificados de
análisis".
Manejo de Reclamos y Retiros del mercado
16.13 Los agentes, corredores, comerciantes,
distribuidores, y reacondicionadores deberán llevar registros de los reclamos y
Retiro del mercado según se especifica en la sección 15.
16.14 Si la situación lo justifica, los agentes, corredores,
comerciantes, distribuidores, y reacondicionadores deberán evaluar el reclamo a
fin de determinar alguna posible acción a iniciar ya sea hacia otros clientes
que pudieran haber recibido el mismo APIs, o hacia las autoridades
regulatorias, o ambos. La investigación sobre la causa del reclamo será llevada
a cabo y documentada por la parte que corresponda.
16.15 Cuando se transfiere un reclamo al elaborador
original, el registro que llevan los agentes, corredores, comerciantes,
distribuidores, y reacondicionadores deberá incluir la respuesta recibida por
parte el elaborador, así como la fecha y la información provista.
Manejo de las Devoluciones
16.16 Las devoluciones deberán manejarse como se detalla
en la sección 14 "Devoluciones". Los agentes, corredores,
comerciantes, distribuidores, y reacondicionadores deberán llevar registro
documentado de las devoluciones de APIs e Intermediarios.
17.- GENERALIDADES
Introducción
17.1 Esta sección apunta a los controles específicos a
realizarse sobre APIs o Intermediarios elaborados por cultivo celular o
fermentación, utilizando organismos naturales o recombinantes, lo cual no ha
sido adecuadamente cubierto en secciones anteriores. No pretende ser tomada
como una sección aislada, en general las normas BPF de las secciones previas
son aplicables en esta también. Nótese que los principios fermentativos de los
procesos "clásicos" para producción de moléculas pequeñas y de los
procesos que utilizan organismos recombinantes o no recombinantes para
producción de proteínas y/o polipéptidos, son los mismos. Sin embargo, el grado
de control será diferente, y esta sección apuntará a esas diferencias. En
general el grado de control sobre procesos biotecnológicos usados para obtener
proteínas o polipéptidos es mayor que sobre fermentaciones clásicas.
17.2 El término "procesos biotecnológicos" se
refiere al uso de células u organismos que han sido generados o modificados
utilizando técnicas de ADN recombinante, o hibridomas u otra técnica para
producir APIs. Los APIs producidos por procesos biotecnológicos normalmente
consisten en sustancias de alto peso molecular, como proteínas y polipéptidos,
para los cuales se da la guía específica en esta sección. Ciertos APIs de bajo
peso molecular como antibióticos, aminoácidos, vitaminas y carbohidratos pueden
también producirse por tecnología de ADN recombinante. El nivel de control para
estos productos es similar al aplicado sobre fermentaciones clásicas.
17.3 El término "fermentaciones clásicas" se
refiere a procesos que utilizan microorganismos tal como existen en la
naturaleza y/o modificados por métodos convencionales (por ejemplo, irradiación
o mutagénesis dirigida) para producir APIs. Estos APIs normalmente son de bajo
peso molecular como antibióticos, aminoácidos, vitaminas y carbohidratos.
17.4 La producción de APIs o Intermediarios a partir de
cultivos celulares o fermentación involucra procesos biológicos tal como el
cultivo de las células y la extracción o la purificación de material a partir
de los organismos vivos. Nótese que puede ser que involucre otros pasos
adicionales que forman parte del proceso de elaboración, como modificaciones
fisicoquímicas. Los materiales de partida usados (medios de cultivo, buffers)
pueden ser una fuente potencial de contaminantes biológicos. Dependiendo de la
fuente, del método de preparación y del uso que se le dará al APIs o
Intermediario, puede ser necesario realizar controles de carga biológica,
contaminación viral y/o endotoxinas durante la elaboración, y monitorear el
proceso en las etapas apropiadas.
17.5 Deberán establecerse controles adecuados en todas las
etapas de elaboración a fin de asegurar la calidad del APIs o Intermediario.
Mientras que esta guía se aplica al proceso a partir del paso de cultivo o
fermentación, los pasos previos (como el desarrollo del banco celular) deberán
llevarse a cabo bajo los apropiados controles del proceso. Esta guía tendrá
aplicación a partir del momento en que un vial de células es retirado del banco
celular para ser usado en elaboración.
17.6 Deberán realizarse controles ambientales y del
equipamiento a fin de minimizar todo riesgo de contaminación. El criterio de
aceptación para la calidad del medio ambiente y la frecuencia de su monitoreo
dependerá de la etapa de producción y de las condiciones en que se lleva a cabo
(Sistema cerrado, abierto o contenido).
17.7 En general, los controles del proceso deben tener en
cuenta:
- Mantenimiento del Banco de Células de trabajo (cuando
corresponda).
- Inoculación y expansión adecuadas del cultivo.
- Control de los parámetros operativos críticos durante el
cultivo o la fermentación.
- Monitoreo del proceso en relación con el crecimiento
celular, la viabilidad y la productividad, cuando éstos correspondan.
- Implementar procedimientos de extracción y purificación
que remuevan células, desechos celulares y componentes del medio, a fin de
proteger el APIs de alteraciones de la calidad y de toda contaminación,
principalmente microbiológica.
- Monitoreo de la carga biológica y, cuando sea necesario,
de niveles de endotoxinas, en las etapas apropiadas de producción.
- La seguridad viral aplicable es la descripta en la ICH Guideline Q5A (Calidad de Productos Biotecnológicos)
17.8 Cuando sea apropiado, deberá demostrarse la remoción
de componentes del medio, proteínas de la célula huésped, impurezas
relacionadas con el proceso o con el producto, y otros contaminantes.
Mantenimiento del Banco Celular y Registros
17.9 El acceso al banco celular deberá limitarse a
personal autorizado.
17.10 El banco celular deberá mantenerse bajo condiciones
de almacenamiento designadas para mantener la viabilidad y evitar la
contaminación.
17.11 Deben llevarse registros del uso de los viales del
banco celular y de las condiciones de almacenamiento.
17.12 Cuando corresponda, el banco celular deberá ser
periódicamente monitoreado a fin de determinar su aptitud para su uso.
17.13 Para una más completa discusión acerca del banco
celular ver la ICH Guideline Q5A (Calidad de Productos Biotecnológicos).
Cultivo Celular y Fermentación
17.14 Cuando sea necesaria la adición aséptica de
sustratos celulares, medio de cultivo, buffers o gases, deberá usarse de ser
posible un Sistema contenido o cerrado. Si la inoculación inicial o
transferencias o adiciones posteriores se realizan en recipientes abiertos,
deberán existir procedimientos y controles a fin de minimizar el riesgo de
contaminación.
17.15 Cuando la calidad del APIs puede afectarse por
contaminación microbiana, la manipulación con recipientes abiertos deberá realizarse
en un flujo laminar u otro ambiente controlado similar.
17.16 El personal deberá estar vestido adecuadamente, y
tomará precauciones especiales en el manipuleo de los cultivos.
17.17 Se deberán monitorear los parámetros operativos
críticos (temperatura, pH, velocidad de agitación, adición de gases, presión) a
fin de asegurar la consistencia con el proceso establecido. Otros parámetros a
monitorear cuando correspondan serán: crecimiento celular, viabilidad y
productividad. Los parámetros críticos podrán variar de un proceso a otro, y
para la fermentación clásica ciertos parámetros pueden no necesitar monitoreo
(como viabilidad celular).
17.18 El equipamiento para cultivo celular deberá ser
limpiado y esterilizado después de cada uso. El equipamiento para fermentación,
cuando sea apropiado deberá ser limpiado y sanitizado o esterilizado.
17.19 Cuando sea apropiado, el medio de cultivo deberá ser
esterilizado antes de ser usado, a fin de proteger la calidad del APIs.
17.20 Deberán existir procedimientos adecuados a fin de
detectar contaminaciones, y establecer las acciones a tomar en dicho caso.
Deberá incluir procedimientos para determinar el impacto de la contaminación
sobre el producto, y para descontaminar el equipo y retornar a las condiciones
de uso para lotes sucesivos. Los microorganismos extraños detectados durante la
fermentación deberán ser identificados, y evaluado su efecto sobre la calidad
del producto. El resultado de esta evaluación se tendrá en consideración para
decidir el destino del producto elaborado.
17.21 Deberán llevarse registros de los eventos de
contaminación.
17.22 Los equipamientos multiproductos (que se utilizan en
varias elaboraciones distintas) deberán sufrir evaluaciones adicionales tras la
limpieza realizada para cambiar de producto, a fin de minimizar el riesgo de
contaminación cruzada.
Extracción, Aislamiento y Purificación
17.23 Los pasos de extracción, ya sean para la remoción de
células o componentes celulares, como para la recolección de componentes
celulares tras la disrupción celular, debe llevarse a cabo en equipos y áreas
designadas a fin de minimizar el riesgo de contaminación.
17.24 Los procedimientos de extracción y purificación que
remueven o inactivan el microorganismo productor, o desechos celulares, o
componentes del medio de cultivo deben ser adecuados de forma de asegurar el
mantenimiento de la calidad del APIs o Intermediario elaborado.
17.25 Todos los equipamientos deberán ser adecuadamente
limpiados y sanitizados después de su uso. Sin embargo podrán elaborarse varios
lotes sucesivos sin limpiar entre ellos si se demuestra que esto no compromete
la calidad del APIs.
17.26 Si se están utilizando Sistemas abiertos, la
purificación deberá realizarse bajo condiciones ambientales apropiadas a fin de
preservar la calidad del producto.
17.27 Si se utilizan equipamientos multiproductos puede
ser necesario implementar controles adicionales.
Etapas de Remoción o Inactivación Viral
17.28 Para mayor información ver la ICH Guideline Q5A (Calidad de Productos Biotecnológicos).
17.29 Las etapas de remoción o inactivación viral son
pasos críticos en algunos procesos y deben llevarse a cabo dentro de sus
parámetros validados.
17.30 Debarán tomarse las precauciones pertinentes para
prevenir la contaminación viral desde los pasos previos a la
remoción/inactivación hacia los pasos posteriores. Por ese motivo, los procesos
en Sistemas abiertos deberán realizarse en áreas separadas y tener diferentes
unidades de ventilación o acondicionamiento del aire.
17.31 No es común que se utilice el mismo equipo en
diferentes pasos de la purificación, sin embargo si esto ocurriera, dicho
equipo deberá ser adecuadamente limpiado y sanitizado antes de su
reutilización. Deberán tomarse las precauciones apropiadas para prevenir
potenciales transferencias de virus desde pasos previos.
18. APIS PARA USO EN ENSAYOS CLINICOS
Generalidades
18.1 No todos los controles de las secciones previas de
esta guía son adecuados para la manufactura de un nuevo APIs que se usará en investigación
durante su desarrollo.
18.2 Los controles usados en la fabricación de APIs para
usar en ensayos clínicos deberán ser consistentes con la etapa de desarrollo
del producto droga que incorporará al API. Los procedimientos de proceso y
testeo deberán ser flexibles para facilitar cambios a medida que aumenta el
conocimiento del proceso y del testeo clínico de un producto droga progresando
desde las etapas pre-clínicas hasta las etapas clínicas. Cuando el desarrollo
de la droga alcanza la etapa en la que el APIs es producido para usarlo en
productos drogas destinado a ensayos clínicos, los fabricantes deberán asegurar
que los APIs son manufacturados en instalaciones aptas, utilizando
procedimientos apropiados de producción y de control para asegurar la calidad
del APIs.
Calidad
18.3 Se deberán aplicar conceptos apropiados de BPF en la
manufactura de APIs para uso en ensayos clínicos con un mecanismo adecuado de
aprobación para cada lote.
18.4 Una unidad de calidad independiente de la producción
deberá establecerse para la aprobación o rechazo de cada lote de APIs para usar
en los ensayos clínicos
18.5 Alguna de las funciones de testeo comúnmente llevadas
a cabo por la o las unidades de calidad puede ser realizada dentro de otras
unidades organizacionales.
18.6 Las mediciones de calidad deberían incluir un sistema
para teste de materias primas, materiales de empaque, intermediarios, y API.
18.7 Todos los problemas de procesamiento y calidad
deberán ser evaluados.
18.8 El etiquetado de APIs destinados al uso en ensayos
clínicos deberá ser adecuadamente controlado y se deberá identificar el
material como destinado a uso en investigación
Equipamiento y Facilidades
18.9 Durante todas las fases del desarrollo clínico,
incluyendo el uso de instalaciones de pequeña escala o laboratorios para
fabricar lotes de APIs para utilizar en ensayos clínicos, los procedimientos
deberán realizarse asegurando que el equipo es calibrado, limpio y adecuado
para el uso proyectado.
18.10 Los procedimientos para el uso de instalaciones
deberán asegurar que los materiales son manipulados de forma que minimice el
riesgo de contaminación y de contaminación cruzada.
Control de Materiales de Partida
18.11 Los materiales de partida utilizados en la
producción de APIs para uso en ensayos clínicos deberán ser evaluados, o
recibidos ya con análisis del proveedor y sujetos a testeo de identidad. Cuando
un material es considerado riesgoso, el análisis del proveedor debería ser
suficiente.
18.12 En algunas instancias, la aptitud de la materia
prima puede ser determinada antes de su uso, basándose en la aceptabilidad de
reacciones en pequeña escala (ej. testeo de uso) más que sobre testeo analítico
solamente.
Producción
18.13 La producción de APIs para uso en ensayos clínicos
deberá ser documentada en los libros de laboratorio, registros de lotes, o por
otro medio apropiado. Estos documentos deberán incluir información sobre el uso
de los materiales de producción, equipamiento, proceso, y observaciones
científicas.
18.14 Los rendimientos esperados pueden ser más variables
y menos definidos que los rendimientos esperados utilizados en los procesos
comerciales. No se prevén investigaciones sobre las variaciones de rendimiento.
Validacion
18.15 Un proceso de validación para la producción de APIs
a ser utilizados en ensayos clínicos es normalmente inapropiado cuando se
produce un solo lote de APIs o cuando el cambio del proceso durante el
desarrollo del APIs hace difícil o inexacta la replicación de un lote. La
combinación de controles, calibración, y donde sea apropiado, un equipo
calificado, aseguran la calidad del APIs durante esta fase del desarrollo
18.16 Los procesos de validación deberán ser conducidos de
acuerdo con la Sección 12 como cuando los lotes son producidos para uso
comercial, aun cuando tales lotes sean producidos en pequeña escala o escala
piloto.
Cambios
18.17 Son previsibles los cambios durante el desarrollo,
en la medida en que se gana conocimiento y la escala de producción aumenta.
Cada cambio en la producción, especificaciones o procedimientos de testeo
deberá ser registrado adecuadamente.
Controles de Laboratorio
18.18 En tanto los métodos analíticos destinados a evaluar
un lote de APIs para ensayos clínicos no puedan aun ser validados, deberán ser
científicamente aptos.
18.19 Debe establecerse un sistema para guardar muestras
de retención de todos los lotes. Este sistema debe asegurar que una cantidad
suficiente de cada muestra de retención queda durante una lapso apropiado
después de la aprobación, terminación o discontinuación de un producto.
18.20 La fecha de vencimiento y re-evaluación tal como se
define en la sección "Fechas de reevaluación y expiración" es
pertinente para los APIs ya existentes utilizados en ensayos clínicos. Para
APIs nuevos, la Sección "Fechas de re-evaluación y expiración"
normalmente no se aplicará en las etapas iniciales de los ensayos clínicos.
Documentación
18.21 Debe ponerse en práctica un sistema para asegurar
que se documenta y está disponible la información obtenida durante el
desarrollo y la manufactura de los APIs a utilizar en ensayos clínicos.
18.22 Se debe documentar apropiadamente el desarrollo e
implementación de los métodos analíticos utilizados para respaldar la
liberación de un lote de APIs para ser utilizado en ensayos clínicos.
18.23 Deberá utilizarse un sistema para resguardar los
registros de producción y control y la documentación. El sistema deberá
asegurar que se resguarden los registros y documentos durante un tiempo
suficientemente largo después de la aprobación, terminación o discontinuidad de
un producto.
GLOSARIO
APIs (Ingrediente farmacológico activo) = Sustancia Droga
Cualquier sustancia o mezcla de sustancias destinada a ser
usada en la manufactura de un producto medicinal y que, cuando es utilizada en
la producción de una droga, se transforma en ingrediente activo del producto
droga.
Tales sustancias están destinadas a proveer actividad
farmacológica u otro efecto directo en el diagnóstico, cura, alivio,
tratamiento, o prevención de enfermedades o para afectar la estructura y
función del cuerpo.
Auxiliares de proceso
Materiales, excluyendo solventes, utilizados como ayuda en
la manufactura de un intermediario o API, que no participan en sí mismos en una
reacción química o biológica (ej. ayuda de filtrado, carbón activado, etc.)
Calibración
La demostración que produce un instrumento o dispositivo
particular, dentro de límites específicos, por comparación con aquellos
producidos por un Estándar de Referencia o sustancia detectable, a través de un
rango apropiado de mediciones
Calificación o aptitud
Acción de probar y documentar que los equipos y sistemas
auxiliares están instalados adecuadamente, trabajan correctamente, y
efectivamente conducen a los resultados esperados. La calificación o aptitud es
parte de la validación, pero las etapas individuales de aptitud por si solas no
constituyen un proceso de validación.
Carga biológica
El nivel y tipo (sea objetable o no) de microorganismos
que pueden estar presentes en las materias primas, materiales de partida APIs,
intermediarios o APIs. La carga biológica puede no ser considerada
contaminación a menos que los niveles hayan excedido o se hayan detectado
organismos objetables definidos.
Contaminación
La introducción indeseada de impurezas de naturaleza
química o microbiológica, o de una sustancia extraña, dentro de un material de
partida, intermediario, o APIs durante la producción, muestreo, embalaje o re
embalaje, almacenamiento o transporte
Contaminación cruzada
Contaminación de un material o producto con otro material
o producto
Control de calidad (QC)
Evaluar o testear que se cumplan las especificaciones
Control del proceso
Chequeos realizados durante la producción, a fin de
monitorear y, en caso de necesitarlo, ajustar el proceso y/o asegurar que el
intermediario o APIs concuerda con las especificaciones.
Criterio de aceptación
Límites numéricos, rangos u otras medidas adecuadas para
la aceptación de los resultados de testeo.
Crítico
Describe un paso del proceso, la condición del proceso,
los requerimientos de testeo u otro parámetro relevante o punto que debe ser
controlado, dentro de un criterio predeterminado, para asegurar que el APIs
cumple con la especificación.
Cuarentena
Estado de materiales aislados físicamente o por otros
medios efectivos, con decisión pendiente acerca de su aprobación o rechazo
subsiguiente.
Departamento de calidad
Una unidad organizacional independiente de la producción,
que cumple las responsabilidades de la Garantía de calidad y del Control de calidad. Puede darse en forma separada en unidades de QA y de QC o reunidos en
un solo individuo o grupo, dependiendo del tamaño y la estructura de la
organización.
Desvío
Alejamiento de una instrucción aprobada o Estándar
establecido.
Elaboración
Todas las operaciones de recepción de materiales,
producción, empaque, reempaque, rotulado, rerrotulado, control de calidad,
despacho, almacenaje y distribución de los APIs y sus controles asociados.
Elaborador contratado
Un elaborador que realiza algún aspecto de la manufactura
para el elaborador original
Especificación
Una lista de pruebas referidas a los procedimientos
analíticos, y de criterios adecuados de aceptación que son límites numéricos,
rangos, u otro criterio para el test que se describe. Establece un conjunto de
criterios con los cuales el material debe cumplir para ser considerado
aceptable para su uso propuesto.
"Conforme a la especificación" significa que el
material, al ser testeado de acuerdo con los procedimientos analíticos
listados, concuerda con los criterios establecidos de aceptación.
Estándar de referencia primario
Substancia que ha sido probada como material auténtico por
un extenso juego de ensayos analíticos, siendo en consecuencia de alta pureza.
Este Estándar puede ser: 1) obtenido de una fuente
oficialmente reconocida, 2) preparado por síntesis independiente, 3) obtenido
de material de producción de alta calidad, existente, o 4) preparado por
purificación adicional de material de producción existente.
Estándar de referencia secundario
Substancia de calidad y pureza establecidas, demostradas
por comparación de un Estándar de referencia primario, utilizada como Estándar
de referencia para análisis de laboratorio de rutina.
Fecha de expiración
La fecha colocada en el contenedor/etiqueta de un API,
designando el tiempo durante el cual el APIs es supuesto que el producto
permanecerá dentro de las especificaciones de su vida útil, si es almacenado en
las condiciones requeridas, y después de la cual no debería ser usada.
Fecha de re-evaluación
La fecha en la que el material debería ser reexaminado
para asegurar que permanece apto para su uso.
Firma (firmado)
Ver definición posterior de firmado
Firmado
El registro del individuo que llevó a cabo una acción
particular o revisión. Este registro puede ser con iniciales, firma completa a
mano, sello personal, o firma electrónica asegurada y autenticada.
Garantía de calidad (QA)
La suma total de las disposiciones organizadas hechas con
el objeto de asegurar que todos los APIs son de la calidad requerida para el
uso que están destinados y que los sistemas de calidad son constantes y
mantenidos.
Impureza
Todo componente presente en el intermediario o API, que no
sea la entidad deseada.
Intermediario
El material producido durante las etapas del proceso de un
APIs que experimenta cambios moleculares adicionales antes de que se transforme
en API. Los intermediarios pueden o no estar aislados (Nota: esta guía sólo se refiere
a aquellos intermediarios producidos después del punto en que la Compañía ha definido como punto en el que comienza la producción del APIs)
Líquidos madre
El líquido residual que resta luego de los procesos de
cristalización o aislamiento. Un líquido madre puede contener materiales no
reactivos, intermediarios, niveles del API y/o impurezas. Puede ser utilizado
para un proceso adicional.
Lote
Una cantidad específica de material producido en un
proceso o serie de procesos de forma que se espera que sea homogéneo dentro de
límites especificados. En el caso de producción continua, un lote puede
corresponder a una fracción definida de la producción. El tamaño del lote puede
ser definido ya sea por una cantidad fija o por la suma producida en un intervalo
fijo de tiempo.
Material
Término general utilizado para denotar materiales de
partida (materiales iniciales, reactivos, solventes) auxiliares del proceso,
intermediarios, APIs y materiales de embalaje y etiquetado
Material de embalaje
Todo material destinado a proteger un intermediario o APIs
durante el almacenaje o transporte.
Material de inicio API
Una materia prima, intermedia o un APIs que es utilizada
en la producción de otro APIs y que es incorporada como fragmento estructural
significativo dentro de la estructura del APIs. Un material de inicio APIs
puede ser un artículo comercial, un material comprado a uno o más proveedores
bajo contrato o acuerdo marcial, o producido domésticamente. Los materiales de
inicio APIs son definidos normalmente por sus propiedades y estructura química.
Materia prima
Término general utilizado para denotar materiales de
inicio, reactivos y solventes destinados a ser utilizados en la producción de
intermediarios o de APIs.
Número de Lote
Una combinación única de números, letras, y/o símbolos que
identifica un lote y por el cual puede ser determinado el historial de la
producción y distribución.
Perfil de impureza
Una descripción de las impurezas identificadas y no
identificadas, presentes en el APIs.
Procedimiento
Descripción documentada de las operaciones a desarrollar,
las precauciones que se deben tomar y las medidas a aplicar directa o
indirectamente, relacionadas con la manufactura de un intermediario o APIs
Producción
Todas las operaciones involucradas en la preparación de un
APIs desde la recepción de los materiales a lo largo de todo el proceso y hasta
el acondicionamiento.
Producto droga (medicinal)
La fórmula de dosificación ya en el envase final
inmediato, destinado al mercado (referencia Q1A)
Protocolo de validación
Un plan escrito mencionando como debe ser conducida la
validación y que define el criterio de aceptación.
Por ejemplo, el protocolo para un proceso de manufactura
identifica el equipo de procesamiento, los rangos críticos de parámetros de proceso
y de operación, características del producto, muestras, datos del test que
deben recogerse, número de corridas de validación, y resultados aceptables del
testeo.
Reelaboración
Someter a un intermediario o API que no conforma los
Estándares o especificaciones a uno o mas pasos del proceso, difiriendo del
proceso de manufactura establecido, para obtener un intermediario o APIs de
calidad aceptable (ej. recristalización con diferente solvente).
Rendimiento esperado
La cantidad de material o el porcentaje de rendimiento
teóricamente esperado, en una fase apropiada de producción basado en escalas
piloto de laboratorio previas, o en datos de fabricación.
Rendimiento teórico
La cantidad que debería ser producida en una fase
apropiada de producción, basada en la cantidad de material utilizado, y en la
ausencia de toda pérdida o error en la producción actual
Reprocesamiento
Introducción de un intermediario o APIs, incluso uno que
no conforme los Estándares o especificaciones, nuevamente en el proceso y
repitiendo un paso de cristalización u otro paso de manipulación química o
física (ej: destilación, filtración, cromatografía, molienda) que sean parte
del proceso establecido de manufactura.
A la continuación de un paso del proceso después de que el
testeo de control del proceso haya demostrado que la etapa está incompleta, se
la considerará parte de un proceso normal, y no un Reprocesamiento.
Sistema computarizado
Un grupo de componentes de hardware y su software asociado,
diseñados y organizados para cumplir una función específica o grupo de
funciones.
Solvente
Liquido orgánico o inorgánico utilizado como vehículo para
la preparación de soluciones o suspensiones en la manufactura de un
intermediario o API
Sustancia droga
Ver APIs (o sustancia droga)
Validación
Programa documentado que suministra un alto grado de
seguridad de que un proceso específico, método, o sistema producirán
consistentemente un resultado que conformará con un criterio predeterminado de
aceptación.
ANEXO VII
BUENAS PRACTICAS DE FABRICACION DE PREPARACIONES
RADIOFARMACEUTICAS
1. ALCANCES
Los presentes lineamientos se encuentran destinados a
complementar aquellos establecidos en las Buenas Prácticas de Fabricación
aplicables a medicamentos y medicamentos estériles.
La regulación aplicable al control de preparaciones o
productos radiofarmacéuticos se encuentra determinada en gran medida, por la
naturaleza de estos productos y los métodos de fabricación.
A los fines de la presente norma, las preparaciones o
productos radiofarmacéuticas se clasifican en:
(a) Productos radiactivos listos para su uso
(b) Generadores de radionucleídos
(c) Componentes no radiactivos ("kits fríos " o
"juegos de reactivos") utilizados en la preparación de compuestos
marcados con un componente radiactivo (generalmente el eluído de un generador
de radionucleídos)
(d) Precursores utilizados en la radiomarcación de otras
sustancias previo a su administración (por ejemplo muestras de pacientes)
2. GENERALIDADES
La fabricación y manipulación de medicamentos
radiofarmacéuticos constituyen operaciones que implican riesgos potenciales
inherentes a la naturaleza de estos productos asociados al tipo de radiación
emitida y a la vida media de los isótopos radiactivos utilizados.
La elaboración de Preparaciones Radiofarmacéuticas debe
ser realizada de conformidad con los principios básicos de Buenas Prácticas de
Fabricación. En particular la elaboración y control de estos medicamentos deben
contemplar las precauciones relacionadas con la radioprotección, prevención de
contaminación cruzada y diseminación de contaminantes radiactivos y la
eliminación de deshechos radiactivos, establecidas en reglamentaciones
nacionales e internacionales.
Las consideraciones contempladas en el presente Anexo
deben ser entendidas como suplementarias a los requerimientos generales de
B.P.F. y específicas para estos productos.
Debido a que algunos preparaciones radiofarmacéuticas son liberadas
y administradas al paciente a poco de su elaboración, el control de calidad
resulta en ciertos casos retrospectivo. Por lo expuesto, el cumplimiento
estricto de B.P.F. resulta imprescindible así como también una evaluación
continua de la eficacia del Sistema de Calidad.
PERSONAL
3.1 Las actividades de elaboración e importación de
productos radiofarmaceúticos debe ser realizada bajo la responsabilidad de un
profesional con formación académica y experiencia demostrada en radiofarmacia y
radioprotrección. El mismo deberá contar con la autorización de la Autoridad Nuclear Competente.
3.2. El personal afectado a las operaciones de fabricación
y manipulación de productos radiactivos debe poseer formación complementaria ya
sea de post grado o mediante entrenamiento técnico, y experiencia apropiada
para dichas funciones. Asimismo deberá recibir información y formación sobre
los aspectos relacionados con la radioprotección.
3.3. El personal afectado al manipuleo de productos
radiactivos o a tareas que deban realizarse en áreas limpias o asépticas debe
ser cuidadosamente seleccionado. A tal fin deberá considerarse su capacidad
para seguir estrictamente los principios de B.P.F. y su estado de salud de
manera tal que la integridad de los productos no se encuentre comprometida .
3.4 La evaluación del estado de salud deberá realizarse
antes del empleo del personal y periódicamente luego de su ingreso. Ante
cualquier alteración del mismo el personal deberá ser separado de actividades
que impliquen su exposición a radiaciones.
3.5. En las áreas Limpias o asépticas sólo deberá estar
presente el personal mínimo necesario para la ejecución del trabajo.
3.6. Durante la elaboración de radiofármacos, juegos de
reactivos o productos estériles, el acceso a estas áreas estará restringido.
Los procedimientos de inspección y control deberán ser realizados, dentro de lo
posible, fuera de estas áreas.
3.7. La movilización del personal entre áreas radiactivas
y no radiactivas sólo podrá realizarse si son respetadas estrictamente normas
de seguridad de radioprotección.
3.8. Deberá establecerse un sistema de capacitación
continua del personal que contemple su entrenamiento en Buenas Prácticas de
Fabricación, manejo seguro de materiales radiactivos y procedimientos de
radioprotección y permita a su vez su acceso al conocimiento de los últimos
desarrollos en los diferentes campos de interés. Deberán ser mantenidos los
registros de la capacitación y realizar una evaluación de la eficacia del
programa de entrenamiento.
3.9. Todo personal involucrado en actividades de
producción, almacenamiento y control de productos radiactivos debe seguir
estrictamente las normas establecidas para el manejo de estos productos y ser
monitoreados por posibles exposiciones a radiaciones y/o contaminación.
4. INFRAESTRUCTURA EDILICIA - EQUIPAMIENTO
4.1 Las instalaciones deben estar localizadas, diseñadas,
construidas y mantenidas conforme a las operaciones que sean realizadas en las
mismas.
4.2. Las áreas donde sean manipulados materiales
radiactivos deberán estar diseñadas teniendo en consideración aspectos
relacionados con la radioprotección, además de aquellos relativos a las
condiciones de Limpieza y esterilidad, cuando corresponda.
4.3. De acuerdo al riesgo radiológico las áreas de
clasificarán en controladas, supervisadas y de libre circulación debiendo estar
definidos los requisitos de acceso.
4.4. Las superficies internas (pisos, paredes y techos) no
deben desprender partículas, deben ser lisas, impermeables y libres de grietas
y permitir su fácil limpieza y decontaminación.
4.5. Debe disponerse de sistemas específicos para la
eliminación de efluentes radiactivos. Estos sistemas deben ser efectivos y
cuidadosamente mantenidos de manera de prevenir contaminación o exposición del
personal a deshechos radiactivos tanto dentro como fuera de las instalaciones.
Deben tomarse las precauciones necesarias para evitar
contaminación del sistema de drenaje con efluentes radiactivos.
4.6. Todas las instalaciones y áreas deben encontrarse en
buen estado de conservación y limpieza, realizándose revisiones regulares y
reparaciones cuando y donde resulte necesario.
4.7. Las instalaciones deben proveer suficiente espacio
para llevar a cabo las operaciones permitiendo un eficiente flujo de trabajo y
una comunicación y supervisión efectiva. Todas las instalaciones deben
encontrarse limpias, en condiciones sanitarias y libre de contaminación
radiactiva.
4.8. La iluminación, sistemas de calefaccionamiento y
ventilación, y de resultar necesario de acondicionamiento de aire, debe estar
diseñado para mantener una temperatura satisfactoria y humedad relativa que
aseguren el confort del personal que deba trabajar con vestimenta protectora.
4.9. El sistema de ventilación de las áreas productivas de
preparaciones radiofarmacéuticas debe cumplir con los requerimientos para la
prevención de contaminación de los productos y la exposición del personal a la
radiactividad.
4.10. Los sistemas de aire, tanto el correspondiente a las
áreas radiactivas como a las no radiactivas, deben estar provistos de alarmas
que permitan advertir al personal sobre posibles fallas del sistema.
4.11. Las áreas de producción y fraccionamiento deberán
contar con blindajes y visores blindados.
4.12. La elaboración de productos radiofarmacéuticos
derivados de sangre o plasma humano deberán realizarse en áreas segregadas y
con equipos dedicados.
4.13. Las autoclaves utilizadas dentro de las áreas
productivas de preparaciones radiofarmacéuticas deberán estar provistas de la
protección adecuada a fin de minimizar la exposición de los operadores a la
radiación. Inmediatamente luego de su utilización, deberá verificarse la
ausencia de contaminación en las mismas a fin de minimizar la posibilidad de
contaminación cruzada por radiactividad entre productos en los próximos ciclos
de autoclavado.
4.14. A fin de prevenir riesgos por contaminación cruzada,
deberán adoptarse todas o algunas de las siguientes medidas:
(a) Procesamiento y envasado en áreas segregadas
(b) Evitar la fabricación simultánea de más de un producto
radiactivo en el mismo puesto de trabajo a fin de disminuir el riesgo de
contaminación cruzada o sustitución, excepto que se encuentren efectivamente
segregados.
(c) Transferencia de material a través de airlocks,
extracción de aire, cambio de vestimenta y lavado y decontaminación cuidadosa
del equipamiento.
(d) Protección contra riesgos de contaminación por
recirculación de aire no tratado o por reingreso accidental de aire extraído.
(e) Utilización de sistemas cerrados de elaboración.
(f) Prevención de formación de aerosoles.
(g) Utilización de recipientes esterilizados.
4.15. Todos los envases que contengan preparaciones
radiofarmacéuticas, independientemente de su estado dentro del proceso de
manufactura, deberán estar correctamente identificados mediante rótulos
seguros.
4.16. La elaboración de productos estériles debe
realizarse en áreas bajo presión positiva. Por lo general, el material
radiactivo debe ser manipulado en áreas específicamente diseñadas mantenidas
bajo presión negativa.
4.17. La elaboración de productos radiactivos estériles
debe ser llevada a cabo en áreas bajo presión negativa rodeada de un área con
presión positiva, asegurando el cumplimiento de los requisitos en cuanto a la
calidad del aire.
4.18. Deberá disponerse de unidades de manejo de aire
independientes para las áreas radiactivas y áreas no radiactivas. El aire
proveniente de las áreas donde hayan sido manipulados materiales radiactivos
deberá ser eliminado a través de filtros apropiados, verificando su desempeño
periódicamente.
4.19. Las cañerías, válvulas y filtros de venteo deben
estar diseñados de forma tal que permitan la validación de limpieza y
decontaminación.
4.20. Los productos radiactivos deben ser almacenados,
tratados, manipulados acondicionados y controlados en locales separados
destinados a tal fin.
5. PRODUCCION
5.1. Todos los procesos de producción deberán ser
realizados siguiendo procedimientos escritos, llevando los registros
correspondientes. Los mismos deben ser periódicamente revisados y actualizados.
5.2. Todos los registros de producción deben estar
inicialados por el operador y verificado en forma independiente por otro
operador o supervisor.
5.3. Las especificaciones de la materia prima deben
incluir detalles de su fuente, origen y, cuando corresponda, el método de
elaboración y los controles utilizados para asegurar su adecuación para el uso
propuesto. En ciertos casos la liberación del producto terminado se encuentra
condicionada por los resultados satisfactorios obtenidos en los ensayos de los
insumos y materia prima.
5.4. Deberá prestarse especial consideración en la
validación de métodos de esterilización.
5.5. En la preparación del producto radiofarmacéutico es
utilizado una amplia variedad de equipamiento. Cuando se utilicen técnicas
cromatográficas para la preparación y purificación de productos deberá evitarse
la contaminación cruzada radioactiva, por lo general mediante el uso de equipos
dedicados a uno o varios productos marcados con el mismo radionucleido. Deberá
estar definida el período de vida útil de las columnas.
5.6. Deben tomarse recaudos en la limpieza, esterilización
y funcionamiento de los liofilizadores utilizados en la preparación de juegos
de reactivos.
5.7. Debe disponerse de un listado de equipamiento y
dispositivos críticos incluyendo balanzas, estufas de depirogenado, dosímetros,
filtros esterilizantes, etc,
5.8. Estos deben ser calibrados y controlados a intervalos
regulares y verificados diariamente o antes del inicio de la producción, teniendo
en cuenta que un error en la lectura y funcionamiento de los mismos pueden
potencialmente causar un perjuicio al paciente. Los resultados de estos ensayos
deben incluirse en los registros diarios de producción
5.9. Deberá disponerse de equipos y dispositivos
específicos para la medición radiactiva como así también de estandares de
referencia. Para la medición de vida media muy corta deberá contactarse con la Autoridad Nuclear competente para la calibración del equipamiento.
5.10. La elaboración y control de kits reactivos deberá
realizarse según las recomendaciones generales de Buenas Prácticas de
Fabricación aplicables a medicamentos estériles.
5.11. En caso de utilizar gas inerte para el llenado de
los viales el mismo deberá ser filtrado a fin de evitar la contaminación
microbiana.
5.12. El acondicionamiento y transporte de radiofármacos
deberá ser realizado siguiendo las normas vigentes en materia de
radioprotección.
6. DOCUMENTACION
6.1. El sistema de documentación deberá seguir los
lineamientos generales contemplados en la B.P.F.
6.2. Los registros para la recepción, el almacenamiento,
uso y descarte de material radiactivo deberán ser mantenidos y llevados
conforme a la reglamentación vigente en materia de radioprotección.
6.3. Los registros de procesamiento de lotes deben incluir
la historia completa de fabricación de cada lote de radiofármaco demostrando
que el mismo ha sido elaborado, controlado, envasado y distribuido de
conformidad con procedimientos escritos.
6.4. Debe llevarse un registro de distribución de todos
los productos, y disponer de procedimientos escritos que indiquen las medidas a
adoptar para el recupero de productos defectuosos liberados al mercado.
6.5. Dado que la devolución de productos radiactivos no
resulta práctica, el objetivo del procedimiento de recupero de estos productos
se encuentra relacionado con la necesidad de prevenir su uso en el paciente en
lugar de lograr el recupero efectivo de los mismos. De resultar necesario, la
devolución de productos radiactivos debe llevarse a cabo de conformidad con las
regulaciones nacionales y/o internacionales en materia de transporte de
material radiactivo.
6.6. El elaborador del producto radiofarmaceutico deberá
poder demostrar que el sistema de recupero adoptado permite llevar la
operatoria eficazmente y dentro de períodos relativamente cortos.
7. ASEGURAMIENTO DE CALIDAD Y CONTROL DE CALIDAD
7.1. Los lotes de productos radiofarmacéuticos con
radionucleídos de período de semidesintegración demasiado corto son por lo
general liberados para su administración antes de la obtención de los
resultados de los ensayos de control de calidad. En estos casos los ensayos
constituyen controles del proceso de elaboración. Por lo expuesto, la
validación del proceso de elaboración empleado resulta crítica y la
implementación y cumplimiento de un Programa de Aseguramiento de la calidad
esencial.
7.2. Las principales responsabilidades de Aseguramiento de
Calidad y/o control de calidad son:
(a) Preparación de instrucciones detalladas para cada
ensayo y análisis.
(b) Asegurar la identificación adecuada y la segregación
de muestras para analizar evitando mezclas, sustituciones o contaminación
cruzada.
(c) Asegurar que el monitoreo ambiental y la validación de
equipos y procesos sean llevados a cabo de manera apropiada a fin de evaluar la
adecuación de las condiciones de elaboración.
(d) Liberar o rechazar materias primas, insumos y
productos intermedios
(e) Aprobar o rechazar material de acondicionamiento y
rotulado
(f) Aprobar o rechazar cada lote de producto terminado.
(g) Evaluar la adecuación de las condiciones bajo las
cuales las materias primas, insumos, producto intermedio y producto terminado
son almacenados
(h) Evaluar la calidad y estabilidad de los productos
terminados y, cuando resulte necesario, de las materias primas y de los
productos intermedios.
(i) Establecer las fechas de vencimiento sobre la base del
período de vida útil y su relación con condiciones específicas de
almacenamiento y un programa de Estabilidad.
(j) Establecer y revisar las especificaciones y los
procedimientos de control.
(k) Asumir la responsabilidad por las muestras de
retención de productos radiofarmacéuticos.
(l) Asumir la responsabilidad para mantener registros
adecuados de distribución de productos radiofarmacéuticos.
7.3. La distribución de productos radiofarmacéuticos de
vida media muy corta, liberados previo a la finalización de todos los controles,
no releva al Profesional Responsable de su obligación de tomar la decisión
sobre la conformidad del lote, debiendo quedar ésta formalmente registrada.
En estos casos deberá disponerse de procedimientos
escritos que describan todas los aspectos relacionados a la producción y al
control de calidad que deben ser considerados, examinados y evaluados previo a
la liberación del lote.
Asimismo deberá existir un procedimiento escrito en el que
se encuentren establecidas las acciones a tomar en caso de obtener resultados
no satisfactorios una vez finalizado los controles de calidad
7.4. Las responsabilidades de Aseguramiento de la Calidad y Control de calidad deben estar organizadas en grupos separados.
7.5. Aseguramiento de la calidad debe incluir el monitoreo
y validación de los procesos productivos.
7.6. Control de calidad deberá ser independiente de
producción y funcionar como una unidad autosuficiente en áreas destinadas a tal
fin.
El laboratorio deberá estar diseñado, instalado y equipado
de manera de poder llevar a cabo todos los ensayos necesarios, llevar los
registros correspondientes y permitir el correcto almacenamiento de muestras y
documentación.
7.7. El elaborador de Preparaciones radiofarmacéuticas
deberá realizar todos los controles cualitativos y cuantitativos establecidos
en las especificaciones de materia prima.
Estos sólo podrán ser reemplazados por un sistema de
certificación del material por parte del proveedor calificado y bajo las
siguientes condiciones:
(a) Existencia de historia de producción confiable
(b) El elaborador o proveedor de la materia prima es
auditado regularmente
(c) Por lo menos un ensayo de identidad es realizado por
el elaborador del producto radiofarmacéutico.
7.8. Los procedimientos de muestreo deben ser adecuados
para el propósito del muestreo, el tipo de controles y la naturaleza del
material a muestrear (por ejemplo tamaño pequeño de lote, contenido radiactivo,
etc.). El procedimiento debe estar descripto en un protocolo escrito.
7.9. Deberán conservarse muestras de referencia de cada
lote de producto intermedio o producto terminado en cantidad suficiente, bajo
condiciones de almacenamiento que permitan repetir los ensayos o verificar los
ya realizados en caso de ser requerido.
Estas muestras deben ser conservadas por períodos
apropiados en conforme al período de semidesintegración del componente
radiactivo involucrado, no siendo esto aplicable para radiofarmacos de vida
media muy corta.
8. ROTULADO
Todos los productos deben encontrarse claramente
identificados mediante rótulos los que deben permanecer en sus envases
respectivos durante todas las etapas productivas y condiciones de
almacenamiento.
ANEXO VIII
Productos Fitoterápicos
1. CONSIDERACIONES GENERALES:
1.1. Los lineamientos expuestos en este anexo están
dirigidas a su aplicación en Productos Fitoterápicos.
1.2. Los elaboradores de Productos Fitoterápicos deberán
ajustarse a estas prácticas y a las expuestas en el cuerpo principal de las
Buenas Prácticas de Fabricación para Elaboradores, Importadores y Exportadores
de Medicamentos.
1.3. Contrariamente a los productos farmacéuticos
convencionales, que están fabricados generalmente a partir de materias primas
sintéticas por medio de técnicas y procedimientos reproducibles de fabricación,
los productos fitoterápicos están preparados a partir de material de origen
vegetal que puede estar sujeto a contaminación y deterioro, de modo que estos
pueden variar en su composición y características.
1.4. El control de las materias primas, el almacenamiento
y el proceso de manufactura asume particular importancia debido a la naturaleza
a menudo compleja y variable de muchos productos fitoterápicos, al número de
principios activos y a la poca cantidad de ellos que se encuentran definidos. A
tal efecto, debe aplicarse un adecuado sistema de aseguramiento de la calidad
en la elaboración y el control de calidad de los productos fitoterápicos.
2. CALIFICACION Y VALIDACION:
2.1. La calificación de equipamiento crítico, la
validación de procesos y el control de cambios son particularmente importantes
en la producción de medicamentos fitoterápicos, de los cuales a menudo no se
conocen los constituyentes responsables de la actividad terapéutica. En este
caso, la homogeneidad del proceso de producción asegura constancia de calidad,
eficacia y seguridad lote a lote.
Ver Anexo II "Validación y Calificación".
3. SANITIZACION E HIGIENE:
3.1. Durante el cultivo, cosecha, recolección y procesado
las materias primas son expuestas a un gran número de contaminantes, en
especial microbiológicos. En relación a reducir esta exposición, es requisito
que el personal encargado del manipuleo del material vegetal y productos
fitoterápicos, tenga un alto grado de higiene personal así como también que
haya recibido un entrenamiento adecuado acerca de los cuidados y
responsabilidades referidas a la higiene.
3.2. El personal debe estar debidamente protegido del
contacto con elementos tóxicos y materiales vegetales potencialmente alergénicos
por medio de una indumentaria adecuada.
3.3. Se debe prestar especial atención a la limpieza y
buen mantenimiento de las áreas de producción y depósito, particularmente
cuando se genera polvo.
4. PERSONAL Y ENTRENAMIENTO:
4.1. El personal involucrado en el proceso de elaboración
o en el control de calidad, debe estar bajo la autoridad de una persona
entrenada y con la suficiente experiencia en el área específica de proceso y
control de calidad de materias primas y productos fitoterápicos. Lo mismo se
aplica para la persona autorizada.
4.2. Con el fin de asegurar una alta calidad en los
productos fitoterápicos, el personal debe tener un adecuado nivel de
entrenamiento en áreas como botánica, fitoquímica y farmacognosia. Se llevarán
registros del entrenamiento y periódicamente se evaluará la efectividad de los
programas de entrenamiento realizados.
5. AUTOINSPECCIONES:
5.1. El equipo de autoinspección debe consistir en
personas expertas en sus campos. Al menos un miembro del equipo debe poseer
particular experiencia en drogas vegetales y en los procesos que se realizan
sobre las mismas en la producción de preparados de drogas vegetales y
medicamentos fitoterápicos.
6. RECLAMOS Y RETIROS DE PRODUCTOS:
6.1. La persona responsable del manejo de quejas y
reclamos, debe poseer experiencia en áreas específicas de control de calidad de
materiales vegetales y productos fitoterápicos. Debe tomarse especial atención
para establecer si el reclamo fue causado por adulteración.
6.2. Los medicamentos fitoterápicos retirados del mercado
deben ser segregados en un área segura, que cumpla los requerimientos
especificados en el subtítulo "Areas de depósito", hasta que se
decida su destino final mediante un procedimiento previamente escrito.
7. INSTALACIONES:
Areas de Depósito
7.1 Debido a que las materias primas de origen vegetal y
los preparados de drogas vegetales son fácilmente degradables, atractivos para
ciertos animales y sensibles a la contaminación microbiana, el correcto
almacenamiento de los mismos asume especial importancia.
7.2 Las materias primas vegetales se deben almacenar en
áreas separadas. El depósito debe estar bien ventilado y equipado de manera de
proteger contra el ingreso de insectos y animales, especialmente roedores. Se
deben tomar medidas eficaces para limitar la diseminación de microorganismos
y/o insectos introducidos con las materias primas y para prevenir la
contaminación cruzada.
7.3 Los envases se deben situar de tal manera que permitan
la libre circulación de aire y faciliten la limpieza. A tal efecto los
contenedores deben ser almacenados separados del suelo y separados entre sí con
el fin de facilitar la limpieza e inspección.
7.4 Para minimizar el riesgo de contaminación, no debe
existir contacto directo entre los materiales y las estanterías o pallets
especialmente si éstos son de madera.
7.5 El almacenamiento de plantas, extractos, tinturas y
otras preparaciones de drogas vegetales puede requerir condiciones especiales
de humedad y temperatura o protección contra la luz; debe asegurarse que estas
condiciones son controladas y monitoreadas.
Areas de Producción:
7.6 Con el propósito de facilitar la limpieza y evitar la
contaminación cruzada, deben tomarse precauciones especiales durante el
muestreo, pesada, y procesos de producción. Se debe contar con instalaciones
dedicadas y/o sistemas de extracción de polvo.
7.7 Los recipientes para desechos, claramente rotulados,
deben permanecer tapados hasta su vaciado y lavado que se realizará
diariamente.
Equipamiento:
7.8 La limpieza del equipamiento utilizado en la
producción de medicamentos fitoterápicos es particularmente importante dada la
cantidad de polvo y material vegetal generados, lo cual puede crear condiciones
favorables para el desarrollo de microorganismos. La utilización de aspiradoras
de polvo y limpieza húmeda son los métodos de elección. Por el contrario, el
uso de aire comprimido y/o cepillado debe eliminarse como método de limpieza,
debido a que estos incrementan el riesgo de contaminación.
7.9 Debe existir un área destinada para la limpieza y
almacenamiento de los equipos y utensillos, separada de las áreas de
producción.
7.10 Las partes de los equipos de producción que entran en
contacto con el producto no deben ser reactivas, ni absorbentes, ni ceder
ningún tipo de material, que pueda influir en la calidad del producto.
Preferentemente, se utilizará equipamiento sin componentes de madera, con la
finalidad de prevenir la contaminación.
8. DOCUMENTACION:
Especificaciones para Materias Primas:
8.1 Sólo puede ser alcanzada una consistente calidad en
los productos fitoterápicos, si las especificaciones la materias primas son
definidas de manera rigurosa y detallada. Por esta razón, además de lo
descripto en Guía general de Buenas Prácticas de Fabricación y Control, las
especificaciones para las materias primas deben incluir lo siguiente:
- Nombre botánico (si es apropiado, el nombre de autores
de la clasificación).
- Detalles del origen de la planta (país de origen, y si
es aplicable métodos de cultivo, época de cosecha, procedimientos de
recolección, cantidad de pesticidas utilizados y fecha, etc.)
- Describir si se utiliza la planta entera o que parte/s
de ella.
- Si la materia prima adquirida es desecada, el método de
secado debe estar especificado.
- Descripción de la materia prima basado en la inspección
macroscópica y microscópica.
- La identificación de la materia prima, que debe incluir,
cuando sea apropiado, la identificación de los componentes activos o de los
marcadores conocidos. A los fines de identificación puede ser utilizado un
ejemplar auténtico de la especie a analizar.
- Valoración de componentes con actividad terapéutica
conocida o marcadores. Deben especificarse los límites de aceptación.
- Determinación de posible contaminación con pesticidas y
límites aceptables para tal contaminación.
- Resultados de análisis para la determinación de metales pesados
y posibles contaminantes, especificando los límites de aceptación, como también
materiales extraños y adulteraciones.
- Resultados de análisis para contaminación microbiana,
incluyendo micotoxinas, y contaminación radiactiva (en especial para materias
primas que han sido irradiadas). Deben especificarse los límites de aceptación.
- Otros análisis (tamaño de partícula, índice de
hinchamiento, solventes residuales en preparados de drogas vegetales, etc.).
8.2 Cualquier tratamiento utilizado para reducir la
contaminación microbiana o la eliminación de insectos debe ser documentado. Se
debe incluir en la documentación detalles del proceso, de los análisis para
determinar el grado de contaminación y de los límites aceptados para los
residuos.
8.3 La expresión cualitativa y cuantitativa de las
sustancias activas en las materias primas y en las preparaciones debe
realizarse de las siguientes maneras:
8.3.1 Materia Prima:
(a) Debe ser indicada la cantidad de droga vegetal; o
(b) La cantidad de droga vegetal se puede expresar como un
rango, correspondiendo a una cantidad definida de componentes con actividad
terapéutica conocida.
Ejemplo:
Nombre del Ingrediente activo: Hojas de Sen.
Cantidad: (a) 900 mg.; o (b) 830-1000 mg, correspondiendo
a 25 mg de glucósidos hidroxiantracénicos calculados como Senosido B.
8.3.2. Preparaciones de drogas vegetales:
(a) Debe ser indicada la cantidad equivalente o el
cociente entre la cantidad de droga vegetal y la preparación (esto no se aplica
a los aceites esenciales o fijos); o
(b) La cantidad de preparación se puede expresar como un
rango, correspondiendo a una cantidad definida de componentes con actividad
terapéutica conocida. Ver ejemplo en 9.1.5.1.
8.4 Debe indicarse la composición de cualquier solvente o
mezcla de solventes utilizados, como también el estado físico del extracto.
8.5 Si cualquier otra sustancia o mezcla de sustancias son
agregadas durante el proceso de fabricación de la preparación, estas deben
estar descriptas como "otros ingredientes".
8.5.1 Ejemplo:
Nombre del primcipio activo: Hojas de Sen.
Cantidad: (a) 125 mg de extracto etanolico seco (8:1) o
125 mg de extracto etanólico, equivalente a 1000 mg de Hojas de Sen; o (b) 100-130
mg de extracto etanólico (8:1), correspondiendo a mg de glucósidos
hidroxiantracénicos, calculados como Senósido B.
Otros ingredientes: Dextrina 20-50 mg.
Especificaciones para Productos Terminados:
8.6 Los análisis de control de calidad y las
especificaciones de producto terminado deben ser tales que permitan la
determinación cualitativa y cuantitativa de los ingredientes activos. Si se
conoce la actividad terapéutica de los componentes, éstos deben especificarse y
determinarse cuantitativamente. Cuando esto no es posible, las especificaciones
deben estar basadas en la determinación de marcadores.
8.7 Si el producto terminado o las preparaciones contienen
varias materias primas, y la determinación de los componentes activos
individuales no es posible, puede ser determinado el contenido combinado de
varios componentes activos. Debe justificarse la necesidad de tal
procedimiento.
8.8 Las especificaciones para productos terminados deben
incluir lo siguiente:
- Contaminación microbiana y de metales pesados.
- Uniformidad de peso, desintegración, dureza, friabilidad
(para comprimidos y cápsulas), viscosidad (para fluidos).
- Humedad (en caso de formas farmacéuticas sólidas).
- Características organolépticas.
- Identificación.
- Valoración de componentes activos o marcadores.
- Impurezas provenientes de degradación (identificadas o
no, si es apropiado).
Instrucciones de Proceso:
8.9 Las instrucciones de proceso deben enumerar las
operaciones que se realizarán sobre las materias primas, tales como secado,
molienda, tamizado, etc. incluyendo el control de parámetros críticos como
pueden ser temperatura, y métodos para controlar el tamaño de fragmentos o
partículas, entre otros.
8.10 Las instrucciones de procedimientos utilizados para
disminuir la contaminación microbiana o la eliminación de insectos en las
materias primas deben estar disponibles. Las mismas deben incluir los detalles
del proceso junto con métodos para determinar los residuos de los agentes
utilizados con sus límites de aceptación.
9. PRODUCCION:
9.1. Lotes de materias primas provenientes de diferentes
zonas geográficas pueden ser mezclados siempre y cuando se demuestre que la
mezcla será homogénea microscópicamente, macroscópicamente y químicamente, entre
otras. Este procedimiento debe estar documentado.
9.2. Todos los lotes deben estar previamente aprobados por
control de calidad. Los lotes que se encuentran fuera de especificación no
pueden ser mezclados con otros.
10. CONTROL DE CALIDAD:
10.1. El personal dedicado a esta actividad debe tener
particular experiencia en productos de origen vegetal para llevar a cabo los
análisis de identificación y el reconocimiento de presencia fúngica, de
heterogeneidad, y de adulteraciones, etc. sobre las materias primas.
Muestras de Referencia y Estándares:
10.2 En el caso de productos fitoterápicos, un estándar de
referencia puede ser un ejemplo botánico de una planta, una muestra de una
preparación de una droga vegetal (ej. Extracto conocido), una sustancia
químicamente definida, un constituyente con actividad terapéutica conocida,
sustancias marcadores o impurezas conocidas.
10.3 El laboratorio de control de calidad debe poseer
ejemplares auténticos de las especies vegetales, utilizadas en la elaboración,
con el fin de realizar pruebas comparativas. Es importante, que se cuente con
ejemplares utilizados generalmente en las adulteraciones, ya que en ocasiones,
la molienda y el mezclado reducen considerablemente la probabilidad de
reconocimiento de las mismas.
10.4 Si el medicamento fitoterápico no está descripto en
una farmacopea, puede ser utilizada una muestra de herbario estandarizada de
varias plantas enteras o partes de ella.
Muestreo
10.5 Debido al hecho de que las materias primas son, en general,
agregados de plantas individuales y por tal motivo la heterogeneidad es
considerablemente alta, el muestreo estadístico debe ser llevado a cabo por una
persona particularmente experimentada. Cada contenedor debe ser identificado
por su propia documentación.
Estudios de Estabilidad
10.6 No será suficiente determinar la estabilidad de los
componentes con actividad terapéutica conocida o los marcadores, puesto que las
materias primas de origen vegetal o las preparaciones de drogas vegetales se miran
en su totalidad como un principio activo. Por lo tanto, debe demostrarse lo más
acertadamente posible (ej. Por comparación de perfiles cromatográficos) que las
otras sustancias presentes son estables y que su contenido como proporción del
conjunto sigue siendo constante. Es importante la observación de las
características organolépticas y físicas de las muestras a analizar ya que
pueden ser modificadas por la presencia o ausencia de diversas sustancias que
se encuentran por debajo de los límites de detección.
10.7 En los productos compuestos por varias materias
primas, y cuando no es posible determinar la estabilidad de cada componente en
forma individual, esta podrá ser determinada mediante la comparación de
perfiles cromatográficos, métodos de valoración, y otros ensayos
fisicoquímicos.
GLOSARIO
Constituyentes con Actividad Terapéutica Conocida
Sustancias o grupos de sustancias químicamente definidas
de las cuales se conoce que son responsables o contribuyen a la actividad
terapéutica de preparados de drogas vegetales o productos Fitoterápicos.
Droga Vegetal
Plantas enteras o sus partes, molidas o pulverizadas
(flores, frutos, semillas, tubérculos, cortezas, etc.) frescas o secas, así
como los jugos, resinas, gomas, látex, aceites esenciales o fijos y otros
componentes similares, que se emplean puras o mezcladas en la elaboración de
medicamentos fitoterápicos.
Materia Prima
Droga vegetal o su preparación, con o sin actividad
terapéutica, empleada en la fabricación de medicamentos fitoterápicos,
excluyendo los materiales de envase.
Marcador
Constituyente químicamente definido de la droga vegetal,
con o sin actividad terapéutica, de interés para propósitos de control, que
puede servir para calcular la cantidad de droga vegetal o de sus preparaciones
en el producto final. Estos deben determinarse cuantitativamente en las
materias primas.
Medicamentos Fitoterápicos
Los medicamentos definidos de acuerdo con el Artículo 1º
inciso a) del Decreto Nº 150/92, pero que no reúnen los requisitos establecidos
para las especialidades medicinales o farmacéuticas definidas en el inciso d)
del Artículo 1º de dicha norma, y que contengan como principio activo drogas
vegetales puras y/o mezclas definidas de éstas y/o preparados de drogas
vegetales, tradicionalmente usadas con fines medicinales y que no contengan
sustancias activas químicamente definidas o sus mezclas aun cuando fuesen
constituyentes aislados de plantas, salvo en los casos que así se justifiquen.
Nombre Científico
Nombre en latín actualizado de una droga vegetal que
permite ubicarla taxonomicamente según normas internacionales reconocidas. Debe
incluir Género, especie y autor. Cuando corresponda debe incluir Familia y taxa
menores.
Preparados de Drogas Vegetales
Productos obtenidos a partir de drogas vegetales
(tinturas, extractos, digeridos, pulverizados u otros) donde se involucren
procedimientos tales como extracción, destilación, purificación, secado, etc.
Cada preparado se considerará en su totalidad como un principio activo. Los
jugos, resinas, gomas, látex, aceites esenciales o fijos serán considerados
como drogas vegetales de acuerdo al la definición (Art. 2º de la Resolución 144/98). Se excluyen de esta definición a los constituyentes asilados químicamente
definidos.
ANEXO IX
Normas para la identificación por colores de envases de
las drogas de uso anestesiológico y de las soluciones parenterales
1. INTRODUCCION
1.1 Principio: Considerando
que la falta de identificación por color de envases de drogas de uso
anestesiológico y soluciones parenterales constituyen un potencial peligro para
la vida del paciente, es indispensable establecer un sistema que contribuya a
la correcta identificación de las drogas de uso anestesiológico por grupo de
acción.
1.2 El objetivo del presente anexo es disminuir el riesgo
durante los procedimientos de anestesia y de aplicación de soluciones
parenterales mediante la identificación inequívoca de los productos
farmacéuticos utilizados en anestesióloga.
1.3 Todos los envases deben contener la información
requerida según lo indicado en las BPF y cumplir con la Normativa Nacional vigente al respecto.
2. IDENTIFICACION DE ENVASES DE USO ANESTESIOLOGICO
2.1 Todos los envases de las drogas referentes al tema
deben estar identificados con bandas horizontales que cubrirán, al menos, hasta
300º de la circunferencia del envase.
2.2 Dichas bandas deben estar ubicadas una en el extremo superior
y otra en el inferior, situando la leyenda entre las mismas.
2.3 Las bandas anchas medirán 2 milímetros o más y las finas medirán un milímetro. Los intervalos entre ambos tamaños de banda no
serán menores a un milímetro.
2.4 Según la clasificación terapéutica de las drogas, los
envases deben ser identificados con bandas de los siguientes colores:
(a) Color Rojo básico para drogas vasculotrópicas
cardiovasculares (Efedrina, Etilefrina, Metaraminol, Adrenalina, Dobutamina,
Dopamina, Isoprotenerol, Fenilefrina, Noradrenalina)
(b) Color Rojo y Calavera Roja para Procaína al 50 %
(c) Color Verde y Calavera Verde para drogas relajantes
musculares periféricas
(d) Color Negro para los fármacos analgésicos, banda ancha
superior e inferior para los opiodes y sus derivados y bandas finas banda ancha
superior e inferior para los A.I.N.E.. La utilización de banda ancha negra
superior y fina negra inferior en el mismo rótulo y envase para aquellos
fármacos que tengan una combinación de opiodes y A.I.N.E..
(e) Color Blanco y Leyenda Blanca para la Atropina
(f) Color Amarillo para los fármacos hipnóticos
intravenosos (Tiopental Sódico, Propofol, Midazolam, Ketamina y Etomidato)
(g) Color Naranja básico para los fármacos reversores
(Neostigmina, Flumazenil, Naxolona)
(h) Color Celeste será reservado para aquellas drogas
comprendidas en dos o más grupos
2.5 Cualquier otro medicamento que deba estar presente en
el quirófano durante el acto quirúrgico (medicamento de quirófano) que contenga
una droga que produzca una acción terapéutica diferente a las indicada en el
ítem anterior (antibióticos, otros anestésicos locales, agentes hipotensores,
Furosemida, A.I.E., etc.) y que puedan ser identificado con código de colores
estos no podrán ser aplicados en forma de bandas.
2.6 Si en el futuro surgieran nuevas drogas de uso
anestesiológico se ubicaran en el grupo correspondiente con el color asignado
en este anexo.
3. IDENTIFICACION POR COLORES DE LAS SOLUCIONES
PARENTERALES
3.1 Los envases de las soluciones parenterales de gran
volumen deben identificarse, además de la leyenda correspondiente, con color en
su arte.
3.2 Los envases de los productos deben estar identificados
con los siguientes colores:
(a) Púrpura básico para Dextrosa al 5 %. Además del color
púrpura básico, la Dextrosa al 10 %, Dextrosa al 25 % y Dextrosa al 50 % deben
tener agregada, respectivamente, una, dos y tres banda de 2 mm. de ancho.
(b) Azul básico para Solución Fisiológica
(c) Rojo básico para Solución molar de Cloruro de Potasio
(d) Naranja básico para Agua para Inyectables
(e) Negro básico para Solución Ringer
(f) Marrón básico para Solución Ringer con Lactato
(g) Verde básico para Solución molar de Bicarbonato de
Sodio
ANEXO X
Buenas Prácticas de Fabricación de productos medicinales
derivados de la sangre o del plasma humano
1. INTRODUCCION
Los productos medicinales biológicos derivados de sangre o
plasma humanos (hemoderivados) pueden tener como materias primas células o
fluidos, incluidos la sangre o el plasma. Los hemoderivados presentan algunas
características especiales como consecuencia de la naturaleza biológica del
material del que proceden. Así por ejemplo, este material puede estar
contaminado por agentes transmisores de enfermedades, especialmente virus. La
seguridad de estos productos depende por tanto del control de los materiales de
partida, del origen de los mismos y de los procesos de fabricación
subsiguientes, incluida la eliminación e inactivación de los virus.
Salvo afirmación contraria, los capítulos generales de las
Buenas Prácticas de Fabricación así como los relativos a la fabricación de
productos medicinales estériles y a la fabricación de medicamentos biológicos
se aplican también a los hemoderivados.
Puesto que la calidad del producto terminado se ve
afectada por todas las fases de su fabricación, incluida la recolección de
sangre o plasma, todas las operaciones deben por consiguiente efectuarse
conforme a un sistema adecuado de aseguramiento de la calidad y a las normas de
Buenas Prácticas de Fabricación (BPF) vigentes.
El fabricante deberá tomar las medidas necesarias para
evitar la transmisión de enfermedades infecciosas y se aplican los requisitos y
normas de las monografías de la Farmacopea Europea relativos al plasma destinado al fraccionamiento y a los productos medicinales derivados de sangre o plasma
humanos.
2. ASEGURAMIENTO DE LA CALIDAD
2.1 El aseguramiento de la calidad debe abarcar todas las
fases previas a la obtención del producto final, desde la extracción (incluida
la selección de donantes, las bolsas de sangre, las soluciones anticoagulantes
y los reactivos y equipos usados en los ensayos serológicos) al almacenamiento,
el transporte, el procesamiento, el control de calidad y la distribución de
producto terminado.
2.2 Los procedimientos para determinar la idoneidad de las
personas para donar sangre y plasma utilizados como materias primas para la
fabricación de medicamentos y los resultados del tamizaje de sus donaciones
deben ser documentados por el centro de donación y estar a disposición del
fabricante del medicamento.
2.3 El seguimiento de la calidad de los hemoderivados debe
efectuarse a lo largo de todo el procesamiento de tal forma que pueda
detectarse cualquier desviación de las especificaciones de calidad.
2.4 Por regla general, los hemoderivados que hayan sido
objeto de devolución no deben volverse a comercializar.
3. LOCALES Y MATERIAL
3.1 Los locales utilizados para la extracción de sangre o
plasma deben tener dimensiones, estructura y localización adecuadas para
facilitar su funcionamiento, limpieza y mantenimiento. La extracción,
procesamiento y tamizaje de la sangre y el plasma no deben efectuarse en el
mismo sitio. Deben existir instalaciones adecuadas para entrevistar a los
donantes en privado.
3.2 El material de fabricación, extracción y tamizaje debe
ser diseñado, certificado y mantenido conforme a su destino y no presentar
ningún riesgo. Su mantenimiento y calibrado periódicos deben efectuarse y
documentarse de acuerdo con los procedimientos establecidos.
3.3 En la preparación de hemoderivados, deben utilizarse
procedimientos de inactivación o remoción viral y deben tomarse medidas para
evitar la contaminación cruzada entre los productos tratados y los no tratados;
los locales y el material utilizado para los productos tratados deben ser
específicos y distintos de los utilizados para los productos no tratados.
4. EXTRACCION DE SANGRE Y PLASMA
4.1 Es necesario un contrato entre el fabricante de
hemoderivados y el centro de donación o el organismo encargado de la extracción
de la sangre o el plasma.
4.2 Todos los donantes deben ser identificados de forma
inequívoca en la recepción y de nuevo en el momento de la extracción.
4.3 Las etiquetas numeradas de cada donación deben
comprobarse una segunda vez por separado para asegurarse de que el número
figure en las bolsas de sangre, en los recipientes de muestra y en los
registros de donación.
5. TRAZABILIDAD Y MEDIDAS POSTERIORES A LA RECOLECCION
5.1 Aún respetando totalmente la confidencialidad, debe
haber un sistema que permita rastrear el trayecto de cada donación, tanto a
partir del donante como a partir del producto terminado, incluyendo el cliente
(hospital o profesional sanitario). Por regla general, la identificación del
receptor es responsabilidad del cliente.
5.2 Debe establecerse un procedimiento operativo estándar
que describa el sistema de información mutua entre el centro de recolección de
sangre y plasma y la instalación de fabricación y fraccionamiento de forma que
puedan informarse mutuamente si, con posterioridad a la donación:
• se descubre que el donante no cumplía los criterios
sanitarios requeridos para los donantes;
• una donación posterior de un donante que había tenido un
resultado negativo en las pruebas de marcadores virales en ocasiones
anteriores, da positivo para cualquiera de los marcadores virales;
• se descubre que las pruebas de marcadores virales no se
han efectuado según lo establecido en los procedimientos operativos;
• el donante ha contraido una enfermedad contagiosa
causada por un agente potencialmente transmisible a través de productos
derivados del plasma (VHB, VHC, VHA y otros virus de las hepatitis no-A, no-B,
no-C, HIV 1 y 2 y otros agentes a la luz de los conocimientos actuales);
• el donante contrae la enfermedad de Creutzfeldt-Jackob
(ECJ o VECJ);
• el receptor de sangre o de un componente sanguíneo
contrae con posterioridad a una transfusión una infección que implica o que
puede atribuirse al donante.
Los pasos a seguir en caso de que se presente cualquiera
de las situaciones anteriores deben estar establecidos en procedimientos
operativos estándar. La investigación retrospectiva debe referirse a las
donaciones efectuadas al menos seis meses antes de la última donación negativa.
En caso de que se presente cualquiera de las situaciones anteriores, siempre
debe efectuarse una reevaluación de la documentación del donante. Hay que evaluar
cuidadosamente la retirada del lote de que se trate teniendo en cuenta
criterios tales como el agente transmisible implicado, el tamaño de la mezcla
inicial, el tiempo transcurrido entre la donación y la seroconversión, la
naturaleza del producto y su método de fabricación. Cuando haya indicios de que
una donación que haya servido para constituir una mezcla de plasmas estaba
infectada con el VIH o el virus de la hepatitis A, B o C, el caso debe ponerse
en conocimiento de la autoridad sanitaria y debe recabarse la opinión de la
empresa sobre la conveniencia de continuar la fabricación a partir de la mezcla
implicada o de retirar el producto o los productos del mercado.
6. PRODUCCION Y CONTROL DE CALIDAD
6.1 Previamente a su fraccionamiento, las donaciones de
sangre y plasma, deben ser sometidos individualmente a ensayo mediante un
método validado de sensibilidad y especificidad adecuadas, para detectar los
siguientes marcadores de agentes infecciosos:
• HBsAg;
• anticuerpos anti VIH 1 y VIH 2;
• anticuerpos anti VHC.
Si una de estas pruebas da un resultado positivo
repetidamente, la donación no es aceptable.
6.2 Las temperaturas de almacenamiento especificadas para
la sangre, el plasma y los productos intermedios deben verificarse y validarse
tanto en los almacenes como durante el transporte entre los centros de donación
y los locales de los fabricantes, o entre las diferentes instalaciones de
fabricación. Esta disposición se aplica asimismo a la entrega de estos
productos.
6.3 La mezcla inicial de plasma homogéneo (despu(þe(þe
eˆðÇés de la separaci&(þe_(þe____ f_ˆ__ðÇoacute;n del
crioprecipitado) debe ser sometido a ensayo mediante un método validado de
sensibilidad y especificidad adecuadas. Los resultados deben ser negativos para
los siguientes marcadores de agentes infecciosos:
• HBsAg
• anticuerpos anti VIH 1 y VIH 2
• anticuerpos anti VHC
Las mezclas que den resultado positivo confirmado deben
rechazarse.
6.4 Unicamente pueden comercializarse los lotes
procedentes de mezclas de plasma analizados mediante Tecnología de
amplificación de los ácidos nucleicos (TAN), con resultados negativos para el
ARN del VHC, usando un método de ensayo validado de sensibilidad y
especificidad adecuadas.
6.5 Los requisitos de análisis para la detección de virus
u otros agentes infecciosos deben considerarse a la luz de los nuevos
conocimientos sobre agentes infecciosos y a la vista de la disponibilidad de
métodos de análisis adecuados.
6.6 Las etiquetas que figuren en cada unidad de plasma
almacenado para la constitución de mezclas y fraccionamiento deben cumplir las
disposiciones de la monografía de la Farmacopea Europea "Plasma humano para fraccionamiento" y exhibir al menos el
número de identificación de la donación, el nombre y la dirección del centro de
donación o las referencias del servicio de transfusión sanguínea responsable de
la preparación, el número de lote del recipiente, la temperatura de almacenamiento,
el volumen total o el peso del plasma, el tipo de anticoagulante utilizado y la
fecha de extracción.
6.7 Con el fin de minimizar la contaminación biológica o
la introducción de material extraño en el plasma destinado al fraccionamiento,
el descongelamiento y la constitución de la mezcla inicial deben efectuarse en
una área al menos del grado D, vistiendo ropa adecuada, máscaras faciales y
guantes. Los métodos utilizados para la apertura de las bolsas, constitución de
mezclas y descongelamiento deben controlarse regularmente, por ejemplo
analizando la carga biológica.
6.8 Deben existir métodos para distinguir claramente entre
medicamentos o productos intermedios que han sido sometidos a un proceso de
eliminación o inactivación viral de los que no lo han sido.
6.9 La validación de los métodos utilizados para la
eliminación o inactivación viral no debe efectuarse en instalaciones de
producción para evitar cualquier riesgo de contaminación de la fabricación
habitual con los virus utilizados para la validación.
7. CONSERVACION DE LAS MUESTRAS
7.1 Siempre que sea posible, deben almacenarse muestras de
las donaciones individuales para facilitar cualquier procedimiento de
restrospección que pudiera resultar necesario. Por regla general, esta tarea es
responsabilidad del centro de donación. Las muestras de cada mezcla de plasma
deben almacenarse en condiciones adecuadas durante al menos un año tras la
fecha de caducidad del producto terminado con el plazo de validez más largo.
8. ELIMINACION DE LA SANGRE, EL PLASMA O LOS PRODUCTOS INTERMEDIOS RECHAZADOS
8.1 Debe haber un procedimiento operativo estándar para la
eliminación segura y eficaz de la sangre, el plasma y los productos intermedios.
Anexo XI
Buenas Prácticas de Fabricación de productos medicinales
biológicos para uso humano
1. ALCANCE
El propósito de este ANEXO es complementar las pautas
proporcionadas en las Buenas Prácticas de Fabricación para Elaboradores,
Importadores / Exportadores de Medicamentos.
Los procedimientos de fabricación abarcados por este ANEXO
incluyen:
a) El crecimiento de cepas de microorganismos y células
eucarióticas.
b) La extracción de sustancias de tejidos biológicos.
c) Técnicas de ADN recombinante (ADNr).
d) Técnicas de hibridomas.
e) La propagación de microorganismos en embriones o
animales.
Los productos biológicos fabricados por estas metodologías
incluyen vacunas, inmunosueros, antígenos, hormonas, citoquinas, enzimas y
otros productos de fermentación (incluyendo anticuerpos monoclonales y
productos derivados del ADNr).
2. PRINCIPIO
La fabricación de productos biológicos requiere ciertas
consideraciones especiales relacionadas a la naturaleza de los productos y de los
procesos involucrados. La forma en que los medicamentos biológicos son
producidos, controlados y administrados requiere ciertas precauciones
necesarias.
A diferencia de los productos farmacéuticos tradicionales,
que normalmente se fabrican y controlan usando técnicas químicas y físicas con
un alto grado de consistencia, los productos biológicos se fabrican con métodos
que involucran procesos y materiales biológicos, como el cultivo de células o
la extracción de material de organismos vivos. Estos procesos biológicos tienen
una variabilidad intrínseca y por lo tanto no son constantes la gama ni la
naturaleza de los subproductos.
El control de los productos biológicos casi siempre
implica el empleo de técnicas biológicas que tienen una variabilidad mayor que
las determinaciones fisicoquímicas. El control durante el proceso adquiere gran
importancia en la fabricación de productos biológicos porque ciertas
deficiencias tal vez no sean reveladas por las pruebas del producto final.
3. PERSONAL
3.1 Todo el personal empleado en áreas donde se fabrican
medicamentos biológicos (incluyendo aquellos relacionados con las tareas de
limpieza, mantenimiento y control de calidad) debe recibir entrenamiento
específico relativo a los productos que se fabrican en su área. El personal
debe recibir toda información relevante y entrenamiento en higiene y
microbiología.
3.2 Los responsables de Producción y Control de Calidad
deben tener un adecuado conocimiento sobre áreas científicas específicas tales
como bacteriología, biología, biometría, química, medicina, farmacología,
virología e inmunología además de experiencia práctica para ejercer las
funciones de coordinación y gerencia del personal a cargo.
3.3 Para mantener la seguridad del producto debe tenerse
en cuenta el estado inmunológico del personal. Cuando corresponda, todo el
personal involucrado en producción, mantenimiento, cuidado de animales y
ensayos in vivo, debe ser vacunado con las vacunas específicas y someterse a
controles médicos periódicos. Es necesario evitar el riesgo de contaminación de
un lote de producción con agentes infecciosos y la exposición del personal a
agentes infecciosos, toxinas o alergenos potentes. Las visitas no deben
ingresar a las áreas de producción.
3.4 Debe evitarse en las áreas de producción, cualquier
cambio en el estado inmunológico del personal que pueda afectar en forma
adversa la calidad del producto. La producción de vacuna BCG y productos a base
de tuberculinas deberá restringirse a personal cuidadosamente monitoreado por
controles periódicos del estado inmunológico o radiografía de tórax.
3.5 En el transcurso de un día de trabajo el personal no
debe pasar desde áreas donde pueda estar expuesto a microorganismos vivos hacia
áreas donde se manipulan otros productos o diferentes organismos. Si este
pasaje es inevitable, el personal involucrado en tal producción deberá seguir
procedimientos claramente definidos de decontaminación incluyendo cambio de
vestimenta y calzado y de ser necesario ducha.
4. INSTALACIONES Y EQUIPOS
4.1 El grado de control de la contaminación ambiental con
partículas y microorganismos en las áreas de producción debe adaptarse al
producto y al proceso de manufactura que se lleva a cabo en el sector, teniendo
en cuenta además el nivel de contaminación de las materias primas y el riesgo
del producto final.
4.2 El riesgo de contaminación cruzada entre productos
biológicos, especialmente durante los pasos del proceso de manufactura en el
cual se utilizan microorganismos vivos, puede requerir de precauciones
adicionales con respecto a las instalaciones y equipos, tales como el uso de
instalaciones y equipamientos dedicados, producción por campaña y uso de
sistemas cerrados. La naturaleza del producto así como el equipamiento usado
determinarán el nivel de segregación necesaria para evitar la contaminación
cruzada.
4.3 En principio se utilizarán instalaciones dedicadas
para la producción de vacuna BCG y para la manipulación de organismos vivos
usados en la producción de tuberculinas.
4.4 Se utilizarán también instalaciones dedicadas para el
manejo de Bacillus anthracis, Clostridium botulinum y de Clostridium tetani
hasta que se completen los procesos de inactivación.
4.5 La producción en campaña puede ser aceptada para otros
microorganismos esporo-formadores solamente si se demuestra que las
instalaciones son dedicadas a este grupo de productos, que no se procesa más de
un producto al mismo tiempo y que se dispone de métodos validados de
decontaminación, sanitización y limpieza.
4.6 Para productos tales como anticuerpos monoclonales y
productos preparados por técnicas de ADNr, puede aceptarse la producción
simultánea en el mismo área solamente si se utilizan sistemas cerrados de
biofermentación.
4.7 Los pasos de producción posteriores a la cosecha
pueden ser llevados a cabo simultáneamente en la misma área de producción si se
demuestra que se toman las medidas necesarias para prevenir la contaminación
cruzada. Para vacunas muertas y toxoides, tal procesamiento en paralelo solo
puede realizarse después de la inactivación del cultivo o después de la
detoxificación.
4.8 Deben usarse áreas con presión positiva para procesar
productos estériles, pero por razones de contención puede aceptarse presión
negativa en áreas específicas en los puntos de exposición a patógenos.
Donde se use presión negativa o gabinetes de seguridad
para el procesado aséptico de patógenos, estos sectores deben estar rodeados de
una zona estéril de presión positiva.
4.9 Las unidades de filtración de aire deben ser
específicas para el área de procesamiento involucrada y no debe permitirse la
recirculación del aire proveniente de áreas donde se manejan organismos
patógenos vivos.
4.10 El diseño y layout de las áreas de producción y el
equipamiento instalado deben permitir la limpieza y decontaminación efectiva.
Debe validarse la efectividad de los procesos de limpieza y decontaminación.
4.11 El equipamiento usado para la manipulación de
organismos vivos debe diseñarse de forma de mantener los cultivos en estado
puro y no contaminados por fuentes externas durante el proceso.
4.12 Los sistemas de drenaje, válvulas y filtros de venteo
deben estar apropiadamente diseñados para facilitar la limpieza y
esterilización. Debe priorizarse el uso de sistemas de limpieza "in
situ" (clean in place) y esterilización "in situ" (sterilise in
place). Las válvulas de los vasos de fermentación deberán ser totalmente
esterilizables por vapor. Los filtros de venteo deben ser hidrofóbicos y debe
establecerse y validarse la duración de los mismos.
4.13 Los recipientes para contención de líquidos deben
diseñarse y controlarse de forma de demostrar que no hay riesgo de pérdidas de
material.
4.14 Los efluentes que pueden contener microorganismos
patógenos, deben ser efectivamente decontaminados.
4.15 Debido a la variabilidad de los procesos y productos
biológicos, algunos excipientes o ingredientes pueden ser medidos o pesados
durante el proceso de producción (p. ej. buffers). En estos casos pueden
mantenerse en las áreas de producción pequeñas cantidades de estas sustancias
5. LOCALES PARA ANIMALES
5.1 Para la fabricación de productos biológicos se
utilizan algunos animales como por ejemplo monos (vacuna polio), caballos y
cabras (antivenenos de víbora), ratones (vacuna antirábica) y caballos
(gonadotrofina sérica). Además se utilizan animales para el control de calidad
de la mayoría de los sueros y vacunas como por ejemplo vacuna pertussis
(ratones), pirógenos (conejos) y vacuna BCG (cobayos).
5.2 Los requerimientos generales para Bioterios están
dados en la Disposición ANMAT 6344/96. Los locales para animales usados en
producción y control de productos biológicos deben estar separados de las áreas
de producción y control. Debe monitorearse y mantenerse los registros
correspondientes al estado de salud tanto de los animales que van a ser usados
en producción como los que van a ser usados en control de calidad. Debe
proveerse a los empleados que trabajan en estas áreas de vestimenta especial y
sectores para el cambio de ropa.
6. DOCUMENTACION
6.1 Las especificaciones para las materias primas de
origen biológico pueden requerir documentación adicional como fuente, origen y
métodos de fabricación y control (particularmente microbiológico).
6.2 Se requiere también especificaciones escritas para
productos intermedios y graneles de productos biológicos.
7. PRODUCCION
Materias primas
7.1 La fuente, origen y aceptabilidad de las materias
primas debe ser claramente definidas. De ser necesario cuando los ensayos tomen
largo tiempo, puede permitirse el procesamiento de una materia prima antes de
que estén disponibles los resultados de los ensayos. En tales casos, la
liberación del producto final será condicionada a los resultados satisfactorios
de dichos ensayos.
7.2 Cuando se requiera esterilización de las materias
primas, ésta debe llevarse a cabo, de ser posible, por calor. De ser necesario
pueden ser utilizados otros métodos adecuados para la inactivación de material
biológico (p.ej. irradiación)
Banco celular madre y Banco celular de trabajo
7.3 La producción de medicamentos de origen biológico
obtenidos de cultivos microbianos, cultivos celulares y propagación de
embriones o animales debe basarse en el sistema de Banco celular madre y Banco
celular de trabajo, para prevenir los cambios no buscados de las propiedades
bioquímicas que se producen con subcultivos repetidos o generaciones múltiples.
7.4 El número de generaciones (duplicaciones o pasajes)
entre el Banco celular de trabajo y el producto final debe ser consistente con
lo presentado en la documentación de registro del producto. El escalado de los
procesos no debe cambiar esta relación.
7.5 El banco celular madre y el de Trabajo deben estar
adecuadamente caracterizados y probados para contaminantes. Debe demostrarse la
aptitud para su uso a través de la consistencia de las características y
calidad de los sucesivos lotes del producto. Los lotes semillas y bancos de células
deberán ser constituidos, usados y mantenidos de forma de minimizar el riesgo
de contaminación o alteración.
7.6 La preparación del Banco celular madre y del Banco
celular de trabajo debe realizarse en un ambiente adecuadamente controlado para
proteger ambos materiales y de ser necesario al personal que los manipula.
Durante la constitución de un Banco celular madre y un Banco celular de
trabajo, no deben manipularse simultáneamente otros agentes vivos o material
infeccioso (p.ej. virus, líneas celulares, etc) en la misma área o por el mismo
personal.
7.7 Se deben mantener registros de la evidencia de aptitud
y recuperación de los bancos. Los recipientes de almacenamiento deben estar
herméticamente cerrados, claramente identificados y mantenidos a una
temperatura adecuada. Se debe mantener un meticuloso inventario del material
almacenado. Las temperaturas de almacenamiento de los freezers deben ser
continuamente registradas y para los tanques de nitrógeno líquido
apropiadamente monitoreadas. Debe registrarse cualquier desviación de los
parámetros establecidos y cualquier acción correctiva implementada.
7.8 La manipulación de estos materiales debe realizarse
solamente por personal autorizado y bajo supervisión de una persona
responsable. El acceso al material almacenado debe ser restringido y
controlado. Los diferentes bancos de células deben ser mantenidos de forma tal
de evitar confusión o contaminación cruzada. Se recomienda separar los bancos
celulares Madre y de Trabajo y mantener las diferentes alícuotas en diferentes
lugares para minimizar el riesgo de pérdida total.
7.9 Todos los contenedores de células madre o de trabajo y
los lotes semillas deben ser tratados de forma idéntica durante el
almacenamiento. Una vez removidos del almacenamiento, los contenedores no deben
retornar al stock.
Operaciones
7.10 Deberá demostrarse las propiedades de promoción del
crecimiento para todos los medios de cultivo.
7.11 El agregado de materiales o cultivos a los
fermentadores y otros recipientes y la toma de muestras debe realizarse bajo
condiciones cuidadosamente controladas para asegurar que se mantiene la
ausencia de contaminación. En los procedimientos de agregado de materiales o
toma de muestra, debe aplicarse cuidados especiales para asegurar que los
recipientes están conectados correctamente.
7.12 Los procesos de centrifugación y mezclado de
materiales pueden llevar a la formación de aerosoles, por esto es necesario la
contención de estas actividades para prevenir la transferencia de
microorganismos.
7.13 De ser posible los medios deben ser esterilizados
"in situ". Cuando se pueda deberán usarse filtros esterilizantes en
línea para el agregado a los fermentadores de gases, medio, ácidos, álcalis,
agentes antiespumantes etc.
7.14 Debe tenerse una cuidadosa consideración a la
validación de cualquier proceso de remoción o de inactivación viral.
7.15 Cuando se realice un paso de inactivación o remoción
viral durante el proceso de manufactura se deberán tomar medidas para evitar la
recontaminación del material tratado con material no inactivado.
7.16 Para la cromatografía se utiliza una amplia variedad
de equipamientos y, en general ese equipamiento debe ser dedicado a la
purificación de un producto y debe ser esterilizado o sanitizado entre lotes
distintos. Debe evitarse el uso del mismo equipamiento en diferentes pasos de
producción. Deben definirse los criterios de aceptación, vida media, métodos de
sanitización y esterilización de las columnas.
8. CONTROL DE CALIDAD
8.1 Los controles en proceso juegan un rol especialmente
importante para asegurar la consistencia de la calidad de los productos
biológicos. Aquellos controles que son cruciales para la calidad (p. ej.
remoción viral) pero que no pueden ser llevados a cabo sobre el producto final
deben llevarse a cabo en los pasos de producción apropiados.
8.2 Es necesario retener muestras de productos intermedios
en las condiciones de almacenamiento apropiadas y en cantidad suficiente para
permitir la repetición o confirmación de un control de un lote.
8.3 Es necesario el monitoreo continuo de ciertos pasos de
producción tales como la fermentación. Estos datos deberán formar parte de los
registros de producción del lote.
8.4 Cuando se utilizan cultivos continuos deberán tomarse
consideraciones especiales con los requerimientos de control de calidad
relacionados a este tipo de métodos de producción.