Disposición 3683-2011
Sistema de Trazabilidad
de medicamentos que deberán implementar las personas físicas o jurídicas que
intervengan en la cadena de comercialización, distribución y dispensación de
especialidades medicinales incluidas en el Registro de Especialidades
Medicinales.
Bs.
As., 23/5/2011
VISTO
la Ley Nº
16.463, los Decretos Nº 150/92 y sus modificatorios y complementarios, Nº
1490/92 y Nº 1299/97, las Resoluciones (ex MSyAS) Nº
538/98 y (M.S.) Nº 435/11; las Disposiciones ANMAT Nº
7439/99 y Nº 3475/05; y el Expediente Nº 1-47-3058-11-5, del Registro de esta
Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, y
CONSIDERANDO:
Que
resulta imprescindible que los países adopten una actitud proactiva
a los efectos de contrarrestar el impacto negativo que el comercio de
medicamentos ilegítimos representa para la salud de los pacientes y sus
sistemas sanitarios.
Que
en nuestro país dichas acciones fueron desarrolladas por esta Administración
Nacional de Medicamentos, Alimentos y Tecnología Médica —ANMAT— a partir del
año 1997, con la creación del Programa Nacional de Pesquisa de Medicamentos
Ilegítimos —PPMI—.
Que
dicho Programa fue creado con el objeto de combatir un fenómeno que cobró gran
fuerza en esos días: la producción de fármacos destinados al tratamiento de
diversas enfermedades, en locales no habilitados o simples galpones.
Que
de acuerdo con la estructura organizativa del primer nivel operativo de esta
Administración aprobada por Decisión Administrativa Nº 22/03, es
responsabilidad primaria del Instituto Nacional de Medicamentos registrar y
controlar las drogas, medicamentos, reactivos y elementos de diagnóstico,
cosméticos y otros productos de uso y aplicación en medicina humana mediante
estudios farmacotécnicos, biológicos, farmacológicos
básicos y toxicológicos, como así también de las actividades, procesos y
tecnologías que se realicen en función de la elaboración, fraccionamiento,
importación y/o exportación, depósito y comercialización de dichos productos, a
fin de asegurar a la población el consumo y uso de elementos de calidad
comprobada.
Que
entre las acciones a su cargo para cumplir tal cometido se encuentra la de
vigilar la legitimidad de los productos que se comercializan y son de su
competencia, a través del Programa de Pesquisa de Medicamentos Ilegítimos.
Que
en el marco del aludido Programa se comenzaron a realizar tareas de
fiscalización y control de los establecimientos dedicados a la comercialización
de medicamentos, siendo su objetivo inicial la identificación y erradicación de
los medicamentos falsificados de los canales de distribución de medicamentos.
Que
como consecuencia del trabajo de campo realizado, que continúa en la
actualidad, pudo detectarse, además de la existencia de medicamentos
falsificados, otro tipo de irregularidades (productos robados, de contrabando,
sin registro, etc.), que dieron origen al concepto más amplio de medicamentos
ilegítimos, posibilitando asimismo mensurar la magnitud de este flagelo en el
país, y permitiendo controlar la circulación de dichos medicamentos en el
mercado interno.
Que
la extensión de la fiscalización abarcó principalmente, desde un primer
momento, a las droguerías y farmacias en acciones conjuntas con las
jurisdicciones, a efectos de prevenir y actuar específicamente sobre el
circuito de introducción ilegítima de especialidades medicinales al mercado,
con el objetivo fundamental de garantizar que los medicamentos que lleguen al
consumidor cuenten con la autorización sanitaria respectiva y sean de calidad,
seguridad y eficacia comprobadas.
Que
no obstante el universo de acción del Programa, cuyo alcance se extiende al
ámbito nacional, comprende asimismo otros puntos de venta y suministro de
medicamentos, tales como las distribuidoras de medicamentos, botiquines de
farmacias y establecimientos asistenciales públicos o privados, efectuando en
muchos casos inspecciones en forma conjunta con las autoridades sanitarias
jurisdiccionales.
Que
conforme a lo expuesto, puede afirmarse que el funcionamiento del Programa
Nacional de Pesquisa de Medicamentos Ilegítimos ha seguido un modelo de
desarrollo apoyado en un fuerte componente de fiscalización desde sus primeros
años de funcionamiento.
Que
el Programa Nacional de Pesquisa de Medicamentos Ilegítimos (PPMI) que esta
Administración viene desarrollando y aplicando desde el año 1997, ha permitido reducir
significativamente la presencia de medicamentos ilegítimos en la cadena de
comercialización de medicamentos.
Que
en forma concomitante la
Procuración General de la Nación constituyó mediante Resolución M.P. Nº 54/97 una comisión compuesta por representantes del
Ministerio Público Fiscal, que tuvo por objeto realizar las diligencias
necesarias a fin de obtener todos los elementos de juicio que posibilitaran
iniciar ante los señores jueces que resulten competentes las investigaciones
sumariales correspondientes relacionadas con la adulteración de medicamentos y
su posterior comercialización, ratificada por su similar Nº 154/08.
Que
asimismo, con fecha 18 de diciembre de 2008 esta ANMAT suscribió un Convenio de
Cooperación Interinstitucional con la Procuración General
de la Nación, que
tuvo por objetivo fortalecer las acciones de cooperación llevadas a cabo entre
las partes desde la creación del PPMI y de la mencionada Comisión para la Investigación de
hechos ilícitos relacionados con la adulteración de medicamentos, a través de
una acción conjunta y coordinada, capitalizando la experiencia, información y
conocimientos adquiridos por ambas entidades; con posterioridad dicho Convenio
fue protocolizado por la
Procuración General de la Nación mediante Resolución PGN Nº 173/08.
Que
concomitantemente a las acciones implementadas desde el Programa de Nacional de
Medicamentos Ilegítimos se dictó el Decreto Nº 1299/97, que tuvo dos objetivos
primarios: enumerar a los operadores que ocupan un lugar en la cadena de
comercialización, y describir la secuencia lógica de las operaciones
comerciales de cada uno de estos operadores, ambos objetivos tratados con la
intención de facilitar las medidas de seguimiento de las transacciones
comerciales para evitar el ingreso de productos ilegítimos en el mercado.
Que
de la aplicación de las normas del Decreto Nº 1299/97 surge la obligación de
que todas las transacciones económicas y comerciales vinculadas con
especialidades medicinales en el ámbito interjurisdiccional
deben llevarse a cabo exclusivamente entre laboratorios farmacéuticos,
distribuidoras, droguerías y farmacias, todos ellos habilitados por las
autoridades competentes en cada caso.
Que
toda persona física o jurídica que adquiera especialidades medicinales, bajo
cualquier modalidad, debe asegurarse que las mismas provengan de empresas
debidamente habilitadas para la comercialización de dichos productos.
Que
en virtud de lo dispuesto en el artículo 14 del Decreto Nº 1299/97, se dictó la Resolución ex M.S y A.S. Nº 538/98 por la cual
se reguló el funcionamiento de las empresas de distribución de especialidades
medicinales y medicamentos que operen en jurisdicción nacional o con destino al
comercio interprovincial o entre las provincias y la Ciudad Autónoma de
Buenos Aires y que actúen por cuenta y orden de laboratorios elaboradores y/o
importadores de dichas especialidades.
Que
el artículo 2º de la aludida Resolución estableció que las personas físicas y/o jurídicas que realicen las actividades mencionadas en
el artículo 1º "...estarán sujetas a la obtención previa de la
habilitación de sus establecimientos, otorgada por la ADMINISTRACION NACIONAL
DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA (ANMAT). Tales
establecimientos operarán bajo la dirección técnica de un profesional
universitario farmacéutico".
Que
asimismo la referida Resolución encomienda a la ANMAT la tarea de fiscalizar
la actividad que cumplen las empresas y establecimientos de distribución de
medicamentos, en el marco del Decreto Nº 1299/97, dictando a tal efecto las
normas complementarias necesarias.
Que
en este sentido, la
ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y
TECNOLOGIA MEDICA (ANMAT) dictó la Disposición Nº 7439/99, que regula el
funcionamiento de las empresas distribuidoras de medicamentos y de los
operadores logísticos, como asimismo las buenas prácticas de almacenamiento,
distribución y transporte, para asegurar la conservación de los productos, y el
mantenimiento de su calidad hasta su entrega al consumidor, por lo que de
conformidad con las aludidas buenas prácticas, las empresas distribuidoras,
deben contar con rastreo y reconocimiento de cualquier lote de producto después
de su entrega.
Que
asimismo la
Disposición ANMAT Nº 3475/05 ha incorporado al ordenamiento
jurídico nacional el Reglamento MERCOSUR sobre Buenas Prácticas de Distribución
de Productos Farmacéuticos, aprobado por la Resolución GMC Nº
49/02, según el cual las droguerías y distribuidoras deben contar con un
sistema de gestión de calidad que permita la rastreabilidad
de los productos y la reconstrucción de su trayectoria de modo de posibilitar
su localización, tendiendo a un proceso eficaz de intervención, retiro del
mercado y devolución.
Que
se ha evidenciado la imperiosa necesidad de articular acciones conjuntas entre
las distintas instancias jurisdiccionales que tienen a su cargo el registro, la
fiscalización y la vigilancia de los productos destinados a la medicina humana,
de manera de generar estándares similares en todo el país.
Que
la Organización
Mundial de la
Salud (OMS) ha recomendado que cada país cuente con una
autoridad regulatoria que ejerza la rectoría para
llevar adelante las actividades de fiscalización y control de productos, bajo
un marco normativo adecuado que contemple todo el ciclo del producto desde su
desarrollo, autorización, producción y seguimiento post comercialización.
Que
en el marco del PLAN ESTRATEGICO DE FORTALECIMIENTO DE LAS CAPACIDADES DE
REGULACION, FISCALIZACION Y VIGILANCIA A NIVEL NACIONAL Y PROVINCIAL, la ADMINISTRACION NACIONAL
DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA (ANMAT) suscribió Actas Acuerdos
con todas las jurisdicciones provinciales y del Gobierno de la Ciudad Autónoma de
Buenos Aires a los fines, entre otros, de tender a la armonización de los
instrumentos de los sistemas de fiscalización de las distintas jurisdicciones.
Que
en este contexto se torna imprescindible generar pautas y articular
entendimientos que prevengan desvíos y por ende eventuales riesgos por
incumplimientos de las normativas vigentes.
Que
en consecuencia resulta imperioso adoptar medidas que propendan al cumplimiento
regular de las tareas de fiscalización y al mismo tiempo permitan continuar y
profundizar la prevención y combate a este flagelo a fin de reducir la prevalencia de medicamentos ilegítimos en el mercado.
Que
deben, por tanto, profundizarse los instrumentos y procedimientos que permitan
a esta Administración y a las demás autoridades sanitarias del país realizar un
seguimiento confiable de las distintas etapas que atraviesa un medicamento
hasta llegar al usuario o paciente, contando para ello con la participación
activa de todas las jurisdicciones.
Que
ese marco, en uso de las facultades conferidas por el artículo 13 del Decreto
Nº 1299/97, el Ministerio de Salud de la Nación dictó la Resolución Nº 435/11,
por la cual se establece que "las personas físicas o jurídicas que
intervengan en la cadena de comercialización, distribución y dispensación de
especialidades medicinales, incluidas en el Registro de Especialidades
Medicinales (REM) de la ADMINISTRACION NACIONAL DE MEDICAMENTOS,
ALIMENTOS Y TECNOLOGIA MEDICA (ANMAT), deberán implementar un sistema de trazabilidad que permita asegurar el control y seguimiento
de las mismas, desde la producción o importación del producto hasta su
adquisición por parte del usuario o paciente, y que además permita brindar toda
otra información suministrada en la actualidad por el sistema de troquel para
que en forma inmediata asegure su reemplazo".
Que
de conformidad con el artículo 3º de dicha Resolución, "La ADMINISTRACION NACIONAL
DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA (ANMAT) será la autoridad de
aplicación de la presente Resolución, quedando expresamente facultada para
dictar las normas necesarias para la debida implementación del sistema de trazabilidad establecido por la presente Resolución, dentro
de los cuarenta y cinco (45) días hábiles administrativos contados a partir de
su entrada en vigencia, todo ello sin perjuicio del dictado con posterioridad
de las normas modificatorias, aclaratorias e interpretativas que la ANMAT estime oportunas para
el mejor desenvolvimiento del aludido sistema. La ANMAT definirá, entre otros
aspectos, los lineamientos técnicos generales, características y modalidades
del código unívoco, del sistema de trazabilidad y de
la base de datos a que se refieren los artículos anteriores, y un cronograma de
aplicación gradual del aludido sistema, en función del grado de criticidad y
distintas categorías de medicamentos, procurando que las medidas a implementar
no perjudiquen el acceso a los mismos por parte de la población. Respecto de
los aspectos técnicos, la autoridad de aplicación podrá requerir la
colaboración y/o participación y/o celebrar acuerdos (al solo efecto de tales
aspectos) con entidades públicas o privadas de reconocida idoneidad."
Que
en tal sentido, y de conformidad con lo previsto en la Resolución del
Ministerio de Salud Nº 435/11, corresponde determinar un esquema gradual de
implementación de la trazabilidad de las especialidades
medicinales, teniendo en cuenta la disponibilidad de medios y sistemas
tecnológicos, manteniendo las condiciones de accesibilidad de las mismas para
la población.
Que
el Instituto Nacional de Medicamentos y la Dirección de Asuntos Jurídicos ambos de la ADMINISTRACION NACIONAL
DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA han tomado la intervención de su
competencia.
Que
la presente medida se dicta en uso de las facultades acordadas por los Decretos
Nº 1490/92 y Nº 425/10, y por el artículo 3º de la Resolución del
Ministerio de Salud Nº 435/11.
Por
ello,
EL
INTERVENTOR DE LA
ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y
TECNOLOGIA MEDICA
DISPONE:
Artículo 1º — El Sistema de Trazabilidad de medicamentos
que deberán implementar las personas físicas o jurídicas que intervengan en la
cadena de comercialización, distribución, y dispensación de especialidades
medicinales incluidas en el Registro de Especialidades Medicinales (REM) de
esta Administración Nacional en los términos establecidos en el artículo 1º y
siguientes de la Resolución
del Ministerio de Salud Nº 435/11, resultará de aplicación, en una primera
etapa a todas aquellas especialidades medicinales, ya registradas o que en el
futuro se registren, que contengan en su composición los Ingredientes
Farmacéuticos Activos (IFA’s) incluidos en el ANEXO I
que forma parte integrante de la presente, sea como monodroga
o en asociación con cualquier otro u otros IFA’s y en
las formas farmacéuticas establecidas en el citado anexo.
El
Sistema de trazabilidad establecido, deberá
encontrarse implementado para la totalidad de las especialidades medicinales
antes enunciadas, con el alcance y según el cronograma establecido en el ANEXO
II, que forma parte integrante de la presente Disposición.
Art. 2º —
A los efectos de la aplicación e interpretación de la Resolución M.S.
Nº 435/11 y de la presente Disposición, se adoptan las siguientes definiciones:
a)
Distribución: Cualquier actividad de tenencia, abastecimiento, almacenamiento y
expedición de productos farmacéuticos, sea a título oneroso o gratuito,
excluida la entrega al público.
b)
Dispensa o dispensación: provisión al público de especialidades medicinales,
sea a título oneroso o gratuito.
c)
Especialidad Medicinal: Todo medicamento, designado por un nombre convencional,
sea o no una marca de fábrica o comercial, o por el nombre genérico que
corresponda a su composición y expendio, de composición cuantitativa definida,
declarada y verificable, de forma farmacéutica estable y de acción terapéutica
comprobable, registrado en el Registro de Especialidades Medicinales (REM) de la ANMAT.
d)
Laboratorio: empresa titular de Registro de Especialidades Medicinales, sea
como elaborador y/o importador de las mismas, en los términos de la Ley Nº 16.463 y su normativa
reglamentaria.
e)
Empresa de distribución de medicamentos o Distribuidora: establecimiento
dedicado a la distribución de medicamentos que actúa por cuenta y orden de
laboratorios elaboradores y/o importadores de dichos productos en los términos
de la Resolución
ex MS y AS Nº 538/98 o su equivalente en las normativas jurisdiccionales.
f)
Operador logístico: establecimiento dedicado a la distribución de medicamentos
que actúa por cuenta y orden de las distribuidoras en los términos de la Disposición ANMAT
Nº 7439/99 o su equivalente en las normativas jurisdiccionales.
g)
Droguería: establecimiento dedicado a la distribución de medicamentos por
cuenta propia y al por mayor, en los términos de la Ley Nº 17.565 y su normativa
reglamentaria o su equivalente en las normativas jurisdiccionales.
h)
Farmacia: establecimiento dedicado al despacho y venta al público de
medicamentos en los términos de la
Ley Nº 17.565 y su normativa reglamentaria o su equivalente
en las normativas jurisdiccionales.
i)
Establecimiento asistencial: cualquier tipo de establecimiento, sea público o
privado, dedicado al diagnóstico y/o tratamiento de pacientes, tal como
hospitales, sanatorios, clínicas, etc.
j)
Trazabilidad por unidad: Sistema de seguimiento y
rastreo colocado en el empaque (unidad de venta al público), de las
especialidades medicinales que permite reconstruir la cadena de distribución de
cada unidad de producto terminado, individualmente.
k)
Tiempo real: transmisión de datos en línea (on line) en el mismo momento de producido el evento a ser
informado.
Art. 3º — Los
laboratorios titulares de certificados de Registro de las Especialidades
Medicinales alcanzados por el artículo 1º deberán colocar en el empaque de cada
una de las unidades de venta al público, un soporte o dispositivo con capacidad
para almacenar un código unívoco fiscalizado y auditado por la ANMAT, según las
recomendaciones del estándar GS1, que contenga la siguiente información:
a)
Código comercial del producto (Global Trade Item Number - GTIN)
b)
Número de serie
Las
droguerías deberán implementar el sistema de trazabilidad,
cumplimentando todos los requisitos de esta Disposición, de acuerdo con el
cronograma establecido en el Anexo II, identificando unívocamente todos los
productos que contengan los IFA’s enumerados en el
Anexo 1 de la presente, siempre y cuando no estén ya identificados por los
laboratorios titulares de certificados de Registro de Especialidades
Medicinales involucradas en la presente Disposición, en cuyo caso deberán
capturar ese código y mantenerlo en todas sus operaciones.
Las
droguerías que inicien la trazabilidad, deberán
identificar los empaques (unidades de venta al público), con los siguientes
códigos:
a)
GLN (Global Location Number)
de la droguería
b)
Número de serie
Las
droguerías que a la fecha de entrada en vigencia de la presente Disposición
cuenten con un sistema de trazabilidad implementado
para aquellos productos que contengan los IFA’s
incluidos en el Anexo I, deberán adecuarse a lo establecido en la presente
Disposición, transmitiendo la información respectiva a la Base de Datos administrada
por la ANMAT,
prevista en el artículo 8º, todo ello en el plazo de treinta (30) días desde
que el sistema se encuentre operativo.
Para
ingresar las especialidades medicinales al sistema de trazabilidad
las droguerías deberán informar los datos que documenten la compra de las
mismas al laboratorio titular de registro o a la distribuidora que actúa por su
cuenta y orden. Los datos son:
a-
código de laboratorio o distribuidora que realiza la venta,
b-
fecha de la misma,
c-
número de remito y factura,
d-
GTIN seriado,
e-
cantidad de unidades vendidas,
f-
Número de lote
g-
Fecha de vencimiento.
Art. 4º — Los
laboratorios titulares de Registro de Especialidades Medicinales que contengan
los IFA’s enumerados en el Anexo I, las droguerías y
sus directores técnicos, respectivamente, serán responsables por la correcta
utilización de los códigos unívocos fiscalizados y auditados por la ANMAT, y de la información
anexa incorporada a los mismos, como así también por la actuación de los
proveedores de los soportes físicos, en caso de adquisición de los mismos a
terceros.
Art. 5º —
Los laboratorios y las personas físicas o jurídicas que intervengan en la
cadena de comercialización, distribución y dispensación de las especialidades
medicinales alcanzadas por la presente Disposición, deberán, para poder
comercializar, distribuir y dispensar las mismas, sin excepción, contar con los
elementos de hardware y software apropiados para capturar el código unívoco
previsto en el artículo 3º y asociar al mismo los siguientes datos de
distribución:
a)
Número de Lote
b)
Fecha de vencimiento
c)
Código del destinatario (CUIT, Global Location Number-GLN, etc.)
En
el caso de provisión a pacientes, la farmacia o establecimiento asistencial
proveedor deberá consignar que la entrega fue efectuada a un paciente,
omitiendo sus datos personales por cuestiones de reserva y confidencialidad.
Dicha información sólo deberá estar disponible a los efectos de ser necesario
contactar al paciente en caso de prevenir cualquier riesgo a su salud y/o vida,
o realizar el recupero del mercado de unidades que le fueran entregadas.
d)
Domicilio del destinatario (domicilio efectivo de entrega);
e)
Fecha de entrega
f)
Factura y Remito asociados a la operación de distribución en cuestión.
Art. 6º — Cada
establecimiento involucrado en la cadena de producción, y/o importación,
comercialización, distribución y dispensación de las especialidades medicinales
alcanzadas por la presente Disposición deberá implementar un Sistema de trazabilidad en cuya Base de Datos se almacene la
información mencionada en el artículo anterior.
Dichos
Sistemas y Bases de Datos deberán cumplir con los parámetros técnicos
específicos y de seguridad informática que determine la ANMAT, a través de entidades
públicas o privadas de reconocida idoneidad en la materia.
Previamente
a que un establecimiento se incorpore al régimen de la presente Disposición, la ANMAT, por sí o a través de
las entidades idóneas que ella determine, deberá validar las Bases de Datos y
Sistemas informáticos implementados, de modo de prevenir accesos indebidos,
sustracción y/o modificaciones no autorizadas de la información.
Art. 7º —
Los establecimientos que incorporen los soportes o dispositivos físicos para
identificar unívocamente las especialidades medicinales alcanzadas por la
presente disposición, deberán garantizar que el mismo no pueda ser removido sin
dejar una marca evidente en el empaque, que permita advertir que este último ha
sido violado, o que sin haber mediado esta última circunstancia, impida su
lectura por medio electrónico. El medicamento en tales condiciones será
automáticamente considerado como adulterado y dará lugar a la adopción de todas
aquellas medidas preventivas y/o de índole administrativa a que hubiere lugar,
de conformidad con la normativa aplicable, sin perjuicio de las demás acciones
que pudieran corresponder.
Art. 8º —
Los laboratorios titulares de certificados de Registro de Especialidades
Medicinales inscriptas en el REM involucradas en esta Disposición y las
droguerías deberán informar a la
ANMAT, en tiempo real, los códigos unívocos asignados a
productos y sus movimientos logísticos con la fecha correspondiente a cada uno
de ellos, en forma cifrada para el debido resguardo y seguridad de la
información. Esta información será almacenada en un Sistema de Base de Datos
Central administrado por la
ANMAT, el que será operado por las entidades públicas o
privadas, idóneas en la materia que la
ANMAT determine y a la que sólo tendrá acceso el personal
especialmente autorizado por la autoridad máxima de esta ANMAT al sólo efecto
de fiscalizar los movimientos logísticos.
Los
sucesivos establecimientos que intervengan en la cadena de comercialización,
distribución y dispensación deberán informar asimismo los movimientos
logísticos en los que intervengan y sus fechas.
Serán
considerados movimientos logísticos y deberán ser comunicados, sin perjuicio de
otros que pudieran informarse, los que se indican a continuación:
a)
producto en cuarentena;
b)
distribución del producto a un eslabón posterior, aclarando según corresponda
(distribuidora, operador logístico, droguería, farmacia, establecimiento
asistencial);
c)
recepción del producto en el establecimiento;
d)
dispensación del producto al paciente;
e)
código deteriorado/destruido (como resultado de una baja por destrucción o
rotura);
f)
producto robado/extraviado;
g)
producto vencido;
h)
entrega y recepción de producto como "devolución";
l)
reingreso del producto a stock;
j)
producto retirado del mercado;
k)
producto prohibido.
Art. 9º —
El software utilizado por esta Administración y por cada eslabón de la cadena
de comercialización, distribución y dispensación para la implementación del
Sistema de Trazabilidad, deberá contemplar un sistema
de seguridad, restricciones y alertas que:
a)
permita identificar duplicaciones de códigos y errores respecto de la
información de los productos;
b)
no permita la realización de operatorias no autorizadas (como ventas a un
eslabón superior de la cadena de distribución);
c)
no permita la distribución y/o facturación de productos con códigos que no
hayan sido destinados al establecimiento que pretenda efectuar tal
distribución,
d)
verifique la legitimidad de la cadena de distribución;
e)
permita a esta Administración tomar conocimiento en tiempo real de cualquier
irregularidad, anomalía y/o desviación de los códigos unívocos pertinentes;
f)
garantice que ningún establecimiento acceda a información de la cadena de
distribución correspondiente a transacciones de otros establecimientos de las
que no formen parte.
Art. 10. — La
falta de consignación de los datos contemplados en los artículos anteriores por
parte de los laboratorios titulares de certificados de especialidades
medicinales inscriptos en el REM involucradas en la presente Disposición y/o
por parte de los sucesivos eslabones de la cadena de comercialización,
distribución y dispensa, como así también la negativa por parte de ellos a
permitir el acceso a sus Bases de Datos será considerada falta grave a los fines
de la adopción de las medidas previstas en el artículo 7º de la Resolución M.S.
Nº 435/11, y con los alcances establecidos en los artículos 5º y 6º de la
misma.
Art. 11. —
Los laboratorios titulares de certificados de Registro Especialidades
Medicinales cuyos IFA’s se encuentren incluidos en el
Anexo I de la presente Disposición y los sucesivos eslabones de la cadena de
comercialización, distribución y dispensa de aquéllas, que no implementen el
sistema de trazabilidad conforme a la presente, una
vez vencidos los plazos fijados en el Anexo II, no podrán continuar con la
producción y/o importación, distribución y comercialización y dispensa de las
referidas especialidades medicinales con los alcances establecidos en los
artículos 5º, 6º y 7º de la
Resolución M.S. Nº 435/11.
Art. 12. —
Los involucrados en la cadena de distribución, comercialización y dispensación,
usuarios y pacientes que adquieran los medicamentos alcanzados por la presente
Disposición deberán tener la posibilidad de solicitar que se les informe si las
unidades adquiridas son originales y fueron distribuidas a través de la cadena
de distribución legalmente autorizada, sea a través de sistemas de consulta
utilizando medios informáticos o por cualquier otro sistema de comunicación.
Art. 13. —
Invítase a los Gobiernos de las Provincias y de la Ciudad Autónoma de
Buenos Aires en el marco de las Actas Acuerdo oportunamente celebradas con esta
Administración, a adherir a la presente disposición para su aplicación a la
comercialización, distribución y dispensación de medicamentos que se efectúen
en jurisdicción de sus respectivos territorios.
Producida
la adhesión de las autoridades jurisdiccionales al presente régimen, deberá
disponerse lo conducente a fin de que las personas que la Autoridad Sanitaria
Jurisdiccional determine puedan acceder a la información del Sistema y Base de
Datos Central correspondiente a los movimientos logísticos de los
establecimientos habilitados por la misma.
Art. 14. —
La presente Disposición entrará en vigencia a partir del día siguiente al de su
publicación en el Boletín Oficial.
Art. 15. —
Regístrese; comuníquese, publíquese, dése a la Dirección Nacional
del Registro Oficial y archívese. — Carlos Chiale.
ANEXO
I
INGREDIENTES
FARMACEUTICOS ACTIVOS
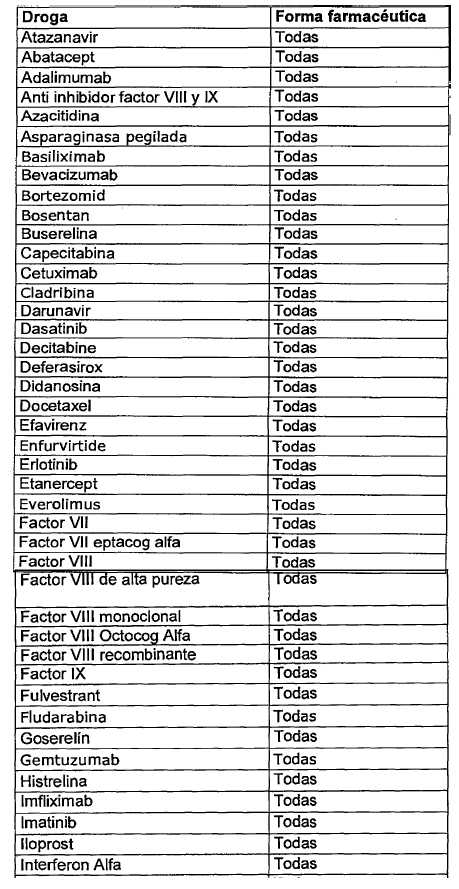
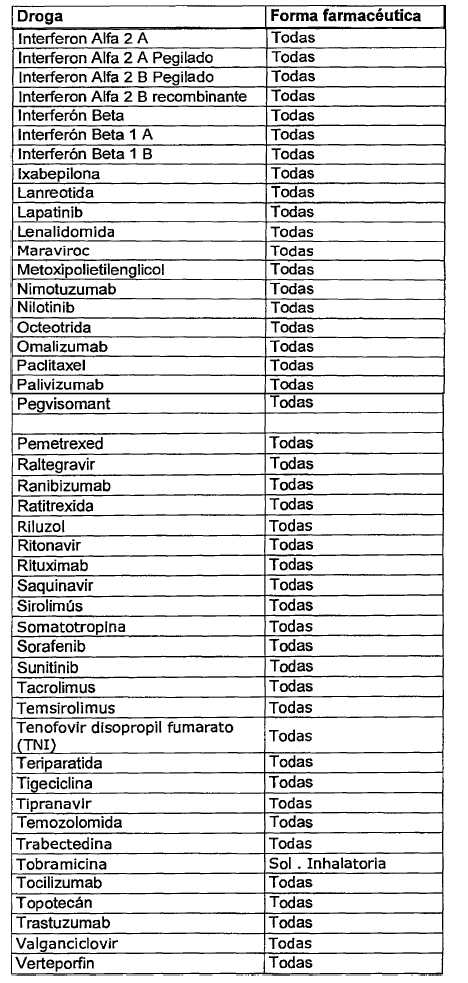
Expediente
n° 1-47-3058-11-5
ANEXO
II
IMPLEMENTACION
DEL SISTEMA DE TRAZABILIDAD PARA PRODUCTOS CRITICOS
ETAPA
1:
•
Desde LABORATORIO hasta DROGUERIA
Hasta
SEIS (6) MESES a partir del 15/06/2011
•
Desde DROGUERIA hasta FARMACIA
Hasta
SEIS (6) MESES a partir del 15/06/2011
ETAPA
2:
VALIDACION
DEL SISTEMA desde:
•
LABORATORIO TITULAR Þ DISTRIBUIDORA Þ DROGUERIA Þ FARMACIA/ESTABLECIMIENTO
ASISTENCIAL Þ PACIENTE
Hasta
DOCE (12) MESES a partir del 15/06/2011